


Abstract
Metoclopramide is increasingly prescribed for conditions previously treated with cisapride, but its metabolic enzymology and drug interactions are poorly understood. Using human liver microsomes (HLMs) and recombinant human cytochromes P450 (P450), we identified the major route of metoclopramide oxidation and the P450 isoforms involved. We also documented the ability of metoclopramide to inhibit the P450 system, using isoform-specific substrate reaction probes of CYP1A2, 2C19, 2C9, 2D6, 2E1, and 3A4. Metoclopramide was predominantlyN-dealkylated to monodeethylmetoclopramide, a metabolite that has not so far been described in humans. Formation rate of this metabolite followed Michaelis-Menten kinetics (Km, 68 ± 16 μM;Vmax, 183 ± 57 pmol/min/mg of protein;n = 3 HLMs). Of the isoform-specific inhibitors tested, 1 μM quinidine was a potent inhibitor of metoclopramide (25 μM) monodeethylation [by an average of 58.2%; range, ∼38% (HL09-14-99) to 78.7% (HL161)] with Kivalues highly variable among the HLMs tested (Ki, mean ± S.D., 2.7 ± 2.8 μM; range, 0.15 μM in HL66, 2.4 μM in HL09-14-99, and 5.7 μM in HLD). Except troleandomycin, which inhibited metoclopramide metabolism in only one HLM (by ∼23% in HL09-14-99), the effect of other inhibitors was minimal. Among the recombinant human P450 isoforms examined, monodeethylmetoclopramide was formed at the highest rate by CYP2D6 (V = 4.5 ± 0.3 pmol/min/pmol of P450) and to a lesser extent by CYP1A2 (0.97 ± 0.15 pmol/min/pmol of P450). The Km value derived (∼53 μM) was close to that from HLMs (68 μM). Metoclopramide is a potent inhibitor of CYP2D6 at therapeutically relevant concentrations (Ki = 4.7 ± 1.3 μM), with negligible effect on other isoforms tested. Further inhibition of CYP2D6 was observed when metoclopramide was preincubated with HLMs and NADPH-generating system before the substrate probe was added (maximum rate of inactivation, Kinact = 0.02 min−1, and the concentration required to achieve the half-maximal rate of inactivation, K′i= 0.96 μM), suggesting mechanism-based inhibition. Metoclopramide elimination is likely to be slowed in poor metabolizers of CYP2D6 or in patients taking inhibitors of this isoform, whereas metoclopramide itself could reduce the clearance of CYP2D6 substrate drugs.
Metoclopramide, since its introduction in the 1960s, is a frequently prescribed drug in adults and children as an antiemetic and for preventing vomiting induced by antineoplastic drugs, particularly cisplatin (Albibi and McCallum, 1983). In addition, it has been widely used as a gastrointestinal (GI1) prokinetic drug to control symptoms of upper GI motor disorders, such as those seen in gastroesophageal reflux disease, dyspepsia, and diabetic gastroparesis (Albibi and McCallum, 1983; Harrington et al., 1983).
Because of the neurological adverse effects of metoclopramide that may occur in up to 20% of patients (Albibi and McCallum, 1983), the use of this drug as a prokinetic has been largely limited after the introduction of cisapride in 1989, a drug that was deemed superior in terms of efficacy and CNS safety (McCallum et al., 1988). However, the use of cisapride has now been restricted in the United States and other countries due to its ability to bring about potentially fatal arrhythmia, leaving a gap in the armamentarium available for treating GI motility disorders. Because no new prokinetic drug is available to replace cisapride, a rapid shift toward the use of older prokinetic agents such as domperidone, erythromycin, and metoclopramide may follow. The increase in the number of deaths due to cisapride, however, raises the disturbing possibility that other prokinetics, including metoclopramide, may have similar effects, in the same way as a number of other antihistamines were shown to be cardiotoxic after the initial reports with terfenadine (Woosley, 1996; Wang et al., 1998). In those studies, metabolic drug interaction was found to be an important risk (Dresser et al., 2000; Michalets and Williams, 2000) as it could be also in the use of domperidone and erythromycin as prokinetic drugs. Domperidone and erythromycin carry proarrhythmic properties (Drici et al., 1998; Drolet et al., 2000), and erythromycin is a known CYP3A inhibitor with the potential of multiple drug interactions (for review, see Dresser et al., 2000).
Because of these concerns, metoclopramide as an alternative to cisapride in adults and children has to be seriously considered, but it too has a questionable safety record. Metoclopramide has been associated with severe adverse effects in the central nervous system, including extrapyramidal symptoms, dystonia, and even death, and there appears to be a correlation between plasma concentrations of metoclopramide and most CNS adverse effects except dystonia (Harrington et al., 1983). Several cases of arrhythmias (heart block, ventricular arrhythmias, and sudden death) with metoclopramide have been documented in the literature (e.g., Shaklai et al., 1974;Bevacqua, 1988; Malkoff et al., 1995; Baguley et al., 1997), and the incidence may increase with wider use of the drug. Furthermore, hypertension (Harrington et al., 1983) and disorders involving hemoglobin oxidation, such as methemoglobinemia (e.g., Wilson et al., 1987; Kearns and Fisher 1988; Grant et al., 1994; Karadsheh et al., 2001) and sulfhemoglobinemia (Van Veldhuizen and Wyatt, 1995; Langford and Sheikh, 1999), have been reported. There is evidence that some of these adverse effects (e.g., dystonia and sulfhemoglobinemia) might be related to the formation of toxic metabolites (Bateman et al., 1983;Van Veldhuizen and Wyatt, 1995), whereas others appear to be exclusively the result of a direct effect of the parent compound (Harrington et al., 1983). As with cisapride and several other drugs, understanding factors that control plasma concentrations of metoclopramide and its metabolites may prove to be helpful in predicting risk.
On the basis of human urinary metabolism data, about 25% of the metoclopramide dose is recovered unchanged, whereas a substantial amount of the dose appears to undergo enzymatic metabolism via oxidation and conjugation reactions (Teng et al., 1977). In animals, the major urinary metabolite was shown to be monodeethylated metoclopramide, which was not identified in humans, and there are nine other putative metabolites found in dogs and rats (Teng et al., 1977). In humans, only one oxidative metabolite, 2-[(4-amino-5-chloro-2-methoxybenzoyl)-amino]acetic acid, has been reported (Teng et al., 1977). The possibility of other pathways is not carefully tested. Indirect evidence from the literature suggests that there may be involvement of the P450 system (Dayer et al., 1992), but the specific enzyme(s) involved in metoclopramide metabolism are not yet known. Although P450-mediated metabolism alone might not determine the overall clearance of metoclopramide, provided that a substantial amount of the dose is excreted unchanged and through conjugation reactions, it can be important in patients whose renal excretion is compromised. Furthermore, a relatively minor pathway might be clinically important for the safety and efficacy of a drug, as has been shown for the metabolism of a number of drugs including procainamide (Lessard et al., 1999), acetaminophen (Nelson, 1990), and codeine (Sindrup et al., 1992).
In the present study, we investigated the interaction between metoclopramide and the P450 system; we characterized the metabolic route of metoclopramide, identified the P450 isoforms involved, and evaluated the ability of metoclopramide to inhibit the P450 system.
Materials and Methods
Chemicals.
Metoclopramide monohydrochloride, dextromethorphan HBr, chlorzoxazone, quinidine sulfate, diethyldithiocarbamate, troleandomycin, phenacetin, sodium acetate, EDTA, MgCl2, G-6-P, G-6-PDH, and NADP were purchased from Sigma-Aldrich (St. Louis, MO). Sulfaphenazole and furafylline were purchased from Ultrafine Chemicals (Manchester, UK). Levallorphan was obtained from the U.S. Pharmacopeia Convention (Rockville, MD). Dextrorphan was purchased from Hoffman-La Roche Inc. (Nutley, NJ). Omeprazole was a generous gift from Dr. Tommy Anderson (Clinical Pharmacology; Astra, Hässle, Mölndal, Sweden). Acetonitrile and triethylamine were purchased from Fisher Scientific (Fair Lawn, NJ).
Human Liver Microsomes and Recombinant P450 Isoforms.
The microsomes used were prepared from human livers that were medically unsuitable for transplantation and frozen at −80°C within 3 h of cross-clamp time. The characteristics of liver donors, procedure for preparation of microsomal fractions, and their P450 contents have been described in our earlier publications (Desta et al., 1998, 2000). The microsomal pellets were resuspended in a reaction buffer (0.1 M Na+ and K+ phosphate buffer, 1.0 mM EDTA, and 5.0 mM MgCl2, pH 7.4) to a protein concentration of 10 mg/ml and were stored at −80°C until used. Protein concentrations were determined with the Bradford method (Bradford, 1976). Other microsomes, as well as baculovirus-insect cell-expressed human P450s 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4, were purchased from Gentest Corp. (Woburn, MA) and stored at −80°C. Protein concentrations and P450 contents were as supplied by the manufacturer.
Incubation Conditions.
To define optimal conditions for incubation and HPLC analysis, metoclopramide (1–100 μM) was incubated in duplicate with HLMs for 0 to 120 min across a range of microsomal protein concentrations (0.1–2 mg of protein/ml) in the presence of NADPH-generating system at 37°C. An incubation time of 60 min and microsomal protein concentration of 1 mg/ml represented linear conditions and were used in subsequent experiments unless otherwise stated. In all experiments, metoclopramide was dissolved and diluted serially with water. Metoclopramide was prewarmed in 1.5-ml Microfuge tubes with sodium monobasic phosphate buffer (pH 7.4) and NADPH-regenerating system (13 mM NADP, 33 mM G-6-P, 33 mM MgCl2, and 0.4 U/ml G-6-PDH) for 5 min at 37°C. Reactions were initiated by adding either 25 μl of microsomes (10 mg of protein/ml) or 20 pmol of recombinant human P450 isoforms and were incubated in a final incubation volume of 250 μl for 60 min at 37°C. Reactions were terminated by placing the incubation tubes on ice, immediately adding 50 μl of ice-cold acetonitrile, and vortex mixing. The samples were then centrifuged at 14,000 rpm for 5 min in an Eppendorf model 5415C centrifuge (Brinkmann Instruments, Westbury, NY). Aliquots of supernatant (120 μl) were drawn off for an injection volume of 100 μl into the HPLC system. Control incubations for each experiment were carried out without substrate, NADPH-generating system, microsomes (using bovine serum albumin instead), or inhibitors.
Assay for Metoclopramide and Its Metabolites.
An HPLC method with UV detection was developed for metoclopramide assay in microsomal incubates. Aliquots of supernatant (100 μl) were injected into the HPLC system. The HPLC system consisted of a Waters (Milford, MA) model 510 or 600 pump, a Waters model 717 autosampler, and a Waters model 996 photodiode array detector or 484 tunable absorbance UV detector. The separation system consisted of an Ace CN column (4.6 × 250 mm, 5-μm particle size; MAC-MOD Analytical, Inc., Chadds Ford, PA), a Waters μBondapak CN guard column (5 μM), and a mobile phase composed of 100 mM sodium acetate/acetonitrile/triethylamine (80:20:0.05 v/v) adjusted to pH 7.0 with acetic acid. The operating temperature was ambient, and the flow rate was 0.9 ml/min. The column eluent was monitored by a photodiode array detector at 274 and 308 nm or by UV detection at 274 nm.
The peak of metoclopramide was noted at a retention time of 14 min, and another major peak was noted at 10 min. This peak was initially designated as metabolite MI. To determine the identity of this metabolite, we analyzed the microsomal incubates of metoclopramide with the help of mass spectrometry and compared the molecular mass of metabolites of metoclopramide that have been so far described in humans and animals. Selected ion recording scans for metoclopramide and its metabolites in HLMs showed MW 271 m/z that corresponds to MI and MW 300 m/z that corresponds to metoclopramide. Based on the molecular mass analysis, MI corresponded to monodeethylmetoclopramide. Subsequent experiments were, therefore, designed to characterize the human P450 isoforms responsible for the formation of MI. Because no synthetic metabolite of MI was available to us, the concentrations of MI were measured comparing the metabolite peak with standard curves obtained with use of known metoclopramide concentrations.
Determination of Kinetics of Metoclopramide Metabolism in HLMs.
Kinetic parameters for the formation of monodeethylmetoclopramide were obtained by incubating a range of metoclopramide concentrations (0.1–150 μM) in duplicate with HLM preparations (or a recombinant human P450 isoform) and an NADPH-regenerating system. The data obtained were fit to the appropriate enzyme kinetic models to calculate the apparent kinetic parameters (Km andVmax) (see data analysis below). These results were compared with those obtained from incubations of metoclopramide over the same concentrations with recombinant human P450 isoforms.
Inhibition of Metoclopramide Metabolism in HLMs.
The formation rate of monodeethylmetoclopramide from 25 μM metoclopramide was evaluated in the absence (control) and presence of the following known isoform-specific inhibitors: furafylline for CYP1A2, sulfaphenazole for CYP2C9, omeprazole for CYP2C19, quinidine for CYP2D6, troleandomycin and ketoconazole for CYP3A, and diethyldithiocarbamate for CYP2E1. These inhibitors are well described in our earlier publications (Desta et al., 1998, 2000). In short, metoclopramide was preincubated for 5 min with and without a P450 isoform-specific inhibitor and with an NADPH-regenerating system. The reaction was initiated by adding HLMs and was incubated for 60 min at 37°C in a final incubation volume of 250 μl. Troleandomycin and furafylline are mechanism-based inhibitors of CYP3A, so they were first preincubated with an NADPH-regenerating system and HLMs for 15 min at 37°C before initiation of the reaction with the addition of metoclopramide. Inhibitors were dissolved in water where appropriate or in a suitable organic solvent (ethanol, methanol, or dimethyl sulfoxide) before being serially diluted with water to contain <0.1% of organic solvents in the final incubation volume. Results of monodeethylmetoclopramide formation were compared with those of controls in which the inhibitor was replaced with the vehicle. Because a marked inhibitory effect was seen in our preliminary experiments with quinidine, with little effects with other isoform-specific inhibitors, Dixon plots were obtained in two different HLMs only for the inhibition of CYP2D6 by quinidine. Metoclopramide (10–50 μM) and quinidine (0–5 μM) were prewarmed for 5 min with an NADPH-regenerating system for 5 min at 37°C. The reaction was then initiated by addition of HLMs and further incubated for 60 min.
Metoclopramide Metabolism by Recombinant Human P450 Isoforms.
To identify specific human P450 isoforms responsible for monodeethylmetoclopramide formation, 25 μM metoclopramide was incubated with 20 pmol each of recombinant P450s 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4. To determine the kinetic parameters in recombinant human CYP2D6, metoclopramide (0.5–150 μM) was incubated with 20 pmol of recombinant proteins. All other incubation conditions remained the same as those for the experiments with HLMs.
Inhibition of P450 by Metoclopramide.
The inhibitory effect of metoclopramide on common drug-metabolizing P450 isoforms was tested in HLMs with isoform-specific substrate reaction probes. Metoclopramide (0.5–100 μM) was incubated in duplicate with HLMs, NADPH regenerating system, and substrate reaction probes specific to each isoform. The reaction probes used were well described in our earlier work (Desta et al., 1998, 2000, 2001). These include phenacetin O-deethylation for CYP1A2, flurbiprofen 4-hydroxylation for CYP2C9, omeprazole hydroxylation for CYP2C19, dextromethorphan O-demethylation for CYP2D6, chlorzoxazone 6-hydroxlation for CYP2E1, and midazolam 1-hydroxylation for CYP3A4. The formation of each metabolite was quantified by calculating the ratio of the area under the curve of the metabolite to the area under of the curve of each internal standard, comparing the ratio with an appropriate standard curve.
To test the potential for mechanism-based inactivation, metoclopramide (25 μM) was preincubated with HLMs and an NADPH-regenerating system for 0 to 30 min at 37°C before the reaction was initiated by the addition of a substrate reaction probe. Since an effect was seen with CYP2D6 and not with any of the other P450 isoforms, further experiments were performed with CYP2D6 to test whether the inactivation was dependent on the time of incubation and inhibitor concentrations. Thus, metoclopramide (0–50 μM) was preincubated for 0 to 30 min before addition of dextromethorphan (25 μM) to test for time and concentration dependence. Dixon plots were obtained in two different HLMs by preincubating metoclopramide (0–25 μM) and the NADPH regenerating system for 20 min at 37°C before initiation of the reaction by addition of dextromethrophan (5–50 μM) and further incubating for an additional 30 min.
Data Analysis.
Apparent kinetic parameters for the formation of monodeethylmetoclopramide (Km andVmax) were calculated using GraphPad (San Diego, CA) Prism software (version 3.02).Km represents substrate concentration at half the maximum velocity (Vmax). In vitro intrinsic clearance is given asVmax/Km. The inhibition (%) of metabolite formation by P450 isoform-specific inhibitors and of P450 substrate probes by metoclopramide was calculated by comparing the inhibited activity with vehicle controls. Initial estimates of inhibitory constants (Ki values) were determined graphically from Dixon plots. Exact Kivalues were then derived from Dixon plots using nonlinear regression for the competitive inhibition model (WinNonLin Software, version 3.1; Pharsight, Mountain View, CA).
Results
Metabolism of Metoclopramide by HLMs.
A typical HPLC chromatogram of metoclopramide and its metabolites (designated as MI and MII) is shown in Fig.1. The major metabolite peak, MI, was formed that depended on NADPH regenerating system, duration of incubation, microsomal protein concentration, and substrate concentration. The retention times for metoclopramide and this major metabolite were 14 and 10 min, respectively. Another minor peak was also noted at 7 min, which may represent a primary or secondary metabolite. Since this metabolite was formed in such small amounts even at high concentrations of metoclopramide (100 μM), no attempt was made to characterize it further.
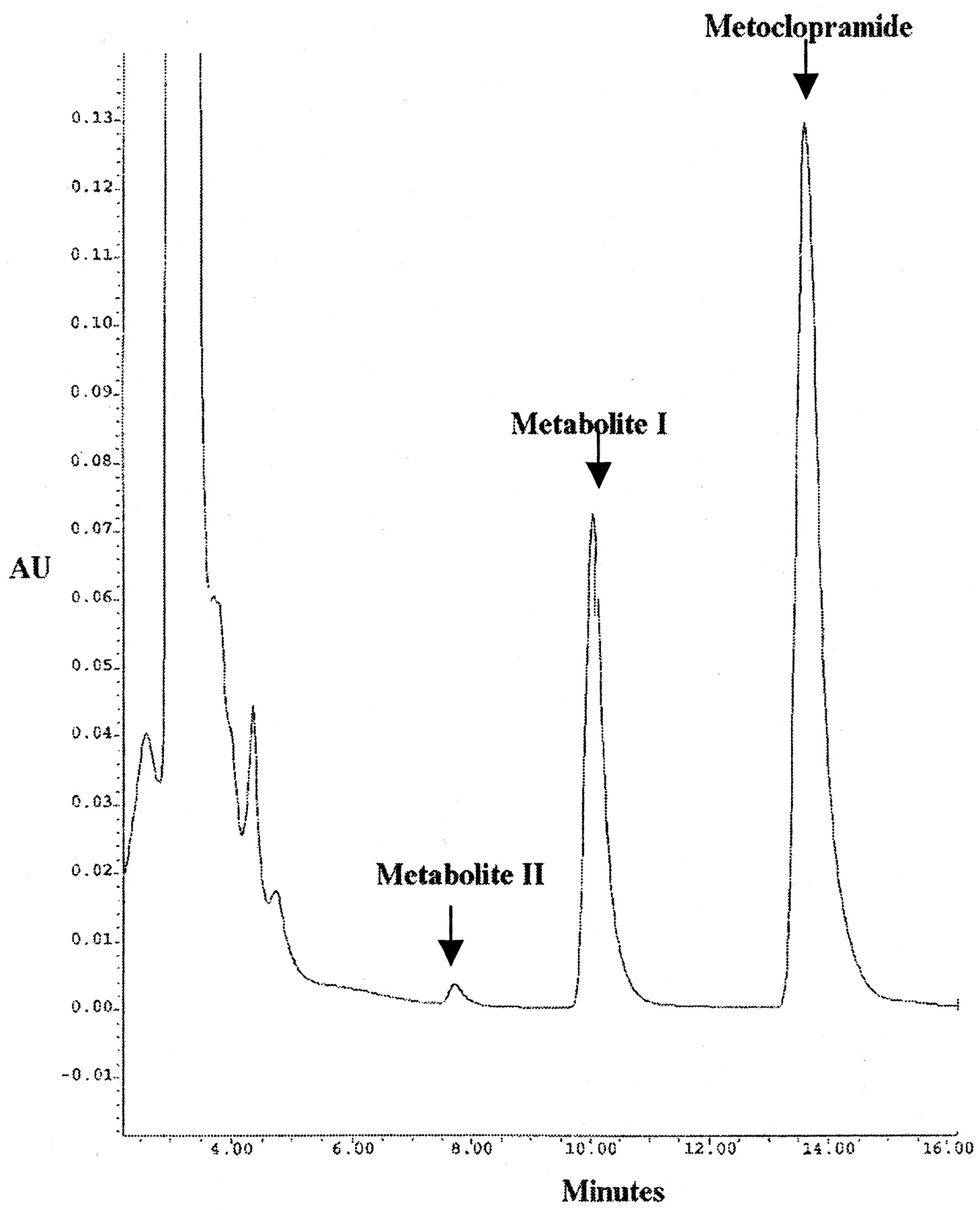
HPLC traces of metoclopramide and its metabolites (MI and MII) from in vitro incubate with recombinant human CYP2D6.
Retention times for metoclopramide, MI, and MII were 14, 10, and 7 min, respectively. For incubation conditions, see Materials and Methods.
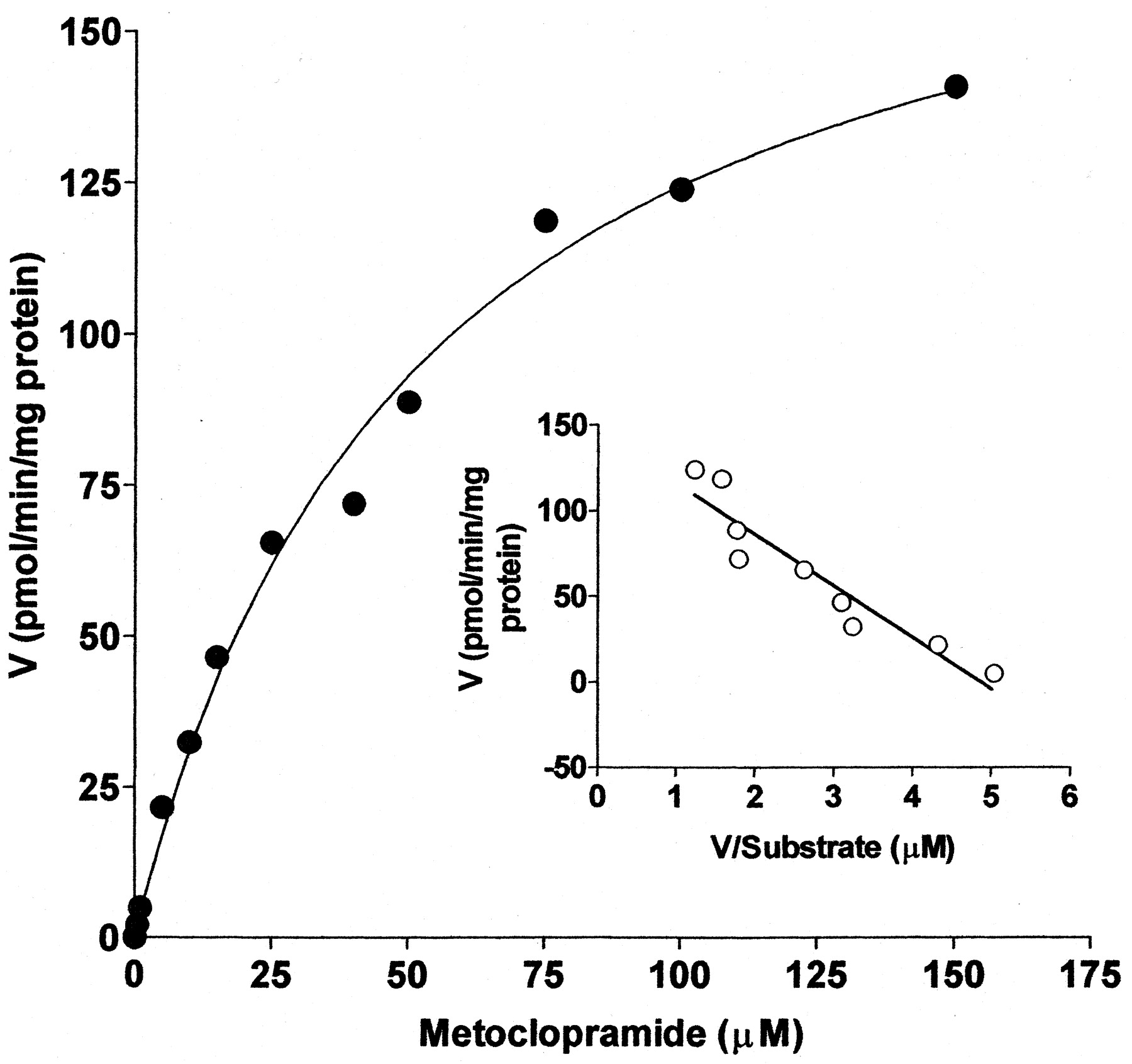
As shown in Fig. 2, the formation of monodeethylated metoclopramide from metoclopramide in HL HK23 followed Michaelis-Menten kinetics. The apparent kinetic parametersKm,Vmax, andVmax/Kmwere derived from three different human livers (Table1). Kinetic parameters for metoclopramide metabolite formation determined in a fourth liver (HL09-14-99) were not included in the calculation of the mean values because it was difficult to estimate them precisely due to what appears to be unsaturable kinetics (Table 1). The insert in Fig. 2 represents the Eadie-Hofstee plot (velocity versus velocity/substrate concentration) and indicates a linear relationship, suggesting involvement of either a single enzyme or more than one enzyme with similar affinity in metoclopramide metabolism.

Michaelis-Menten kinetics for the formation of M1 from metoclopramide (0–150 μM) in HLMs.
Each point represents values obtained after the average of duplicate experimental data was fitted to a one-enzyme Michaelis-Menten model. Eadie-Hofstee plot (velocity versus velocity/substrate concentration) is shown in the inset. For incubation conditions, see Materials and Methods.
Kinetic parameters for the formation of MI from metoclopramide (0–150 μM) in HLMs
Chemical Inhibition of Metoclopramide Metabolism.
To probe the P450 isoforms participating in monodeethylation of metoclopramide, 25 μM substrate was incubated with P450 isoform-specific inhibitors in two different HLMs. As shown in Fig.3 (upper panel), 1 μM quinidine inhibited metoclopramide metabolism by an average of 58.2%. However, it is important to note that the extent of inhibition is highly liver dependent: high in HL161 (by 78.7%) and less in HL09-14-99 (by ∼38%). Whereas the effect of 50 μM troleandomycin (CYP3A-specific inhibitor) on the metabolism of metoclopramide in HL161 was negligible (by <4%), it was a relatively potent inhibitor in HL09-14-99 (by 22.8%) (Fig. 3, upper panel). The effect of multiple quinidine and troleandomycin concentrations on metoclopramide metabolism was tested in this HL09-14-99 (Fig. 3, lower panel). These data suggest that both CYP2D6 and CYP3A are involved in the metabolism of metoclopramide in this particular human liver, with approximately 70 and 30% contribution, respectively. Indeed, the monodeethylation of metoclopramide was completely inhibited (by 93%) when both quinidine (1 μM) and troleandomycin (50 μM) in combination were incubated with metoclopramide in HL09-14-99 (Fig. 3, upper panel, left side). Other isoform-specific inhibitors [sulfaphenazole (CYP2C9), omeprazole (CYP2C19), furafylline (CYP1A2), and diethyldithiocarbamate (CYP2E1)] did not significantly inhibit monodeethylated metoclopramide formation in either liver (Fig. 3, upper panel). The representative Dixon plot for the inhibition of metoclopramide monodeethylation by quinidine is shown in Fig. 4. TheKi values derived from Dixon plots constructed in three different HLMs were highly variable: 0.15 μM in HL66, 2.4 μM in HL09-14-99, and 5.7 μM in HLD (mean ± S.D., 2.74 ± 2.78 μM).
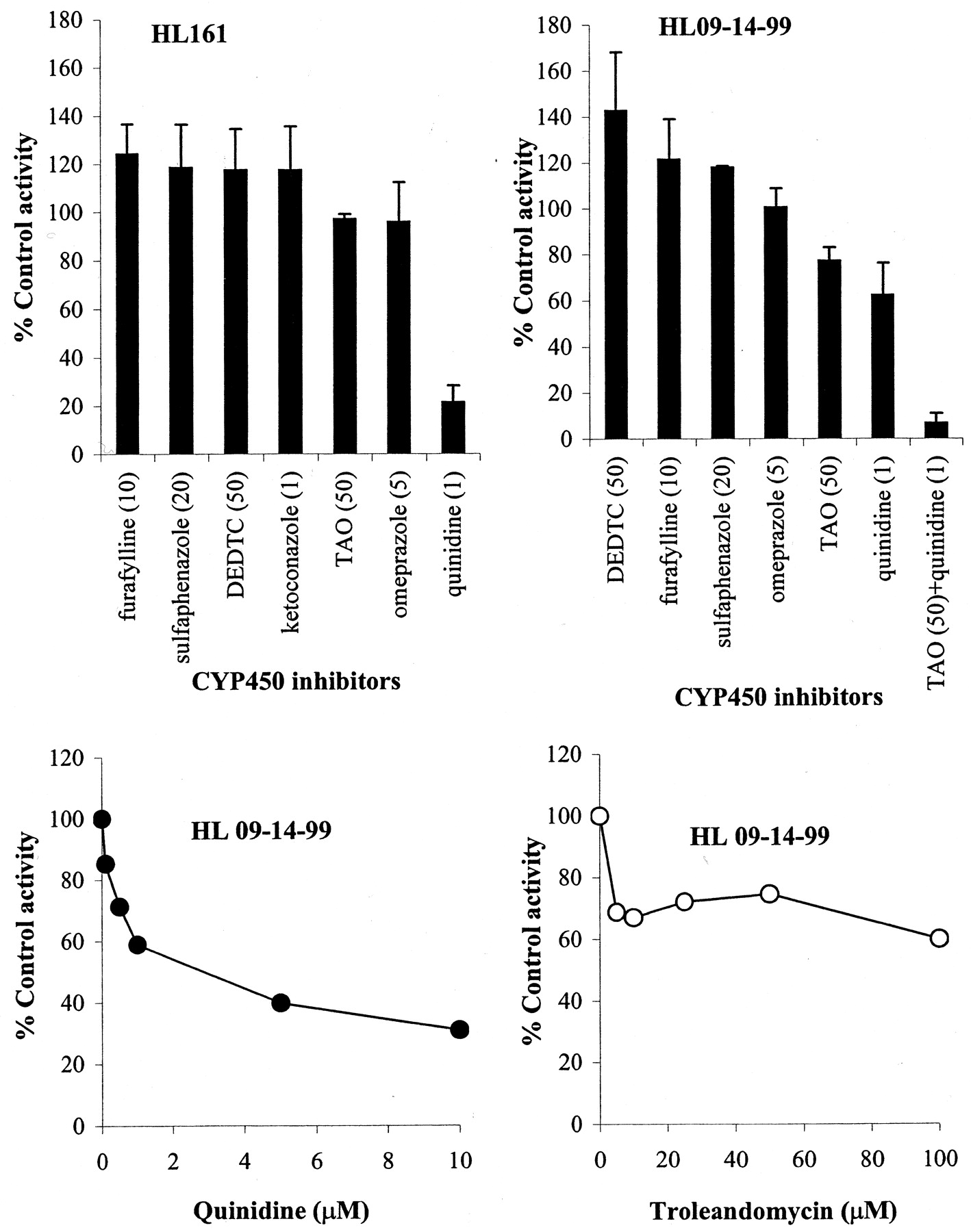
Inhibition of monodeethylation of metoclopramide (25 μM) in two HLMs (HL161 and HL09-14-99) by P450 isoform-specific inhibitors.
The inhibitors used (upper panel) were as follows: furafylline (10 μM) for CYP1A2, sulfaphenazole (20 μM) for CYP2C9, omeprazole (5 μM) for CYP2C19, quinidine (1 μM) for CYP2D6, diethyldithiocarbamate (50 μM) for CYP2E1, troleandomycin (50 μM) and ketoconazole (1 μM) for CYP3A, and a combination of quinidine (1 μM) and troleandomycin (50 μM). Upper panel (left), data from HL161; upper panel (right), data from HL09-14-99; and lower panel, effect of multiple concentrations of quinidine (left) and troleandomycin (right) on metoclopramide (25 μM) metabolism in HL09-14-99. Data represent mean ± S.D. (upper panel) or mean of duplicate measurements (lower panel).

Representative Dixon plots for the inhibition of monodeethylation of metoclopramide (10–100 μM) by quinidine (0–1 μM).
Data are averages of duplicates. For incubation conditions, seeMaterials and Methods.
Metabolism of Metoclopramide by Human Recombinant P450s.
As demonstrated in Fig. 5, production of monodeethylmetoclopramide was catalyzed at the highest rate by CYP2D6 (V = 4.5 ± 0.3 pmol/min/pmol of P450) and CYP1A2 (V = 0.97 ± 0.15 pmol/min/pmol of P450), whereas this turnover was small in recombinant human CYP2C19 (V= 0.24 pmol/min/pmol of P450), CYP3A4 (V = 0.097 ± 0.01 pmol/min/pmol of P450), and CYP2C8 (V = 0.039 ± 0.006 pmol/min/pmol of P450). The other isoforms (CYP2A6, 2B6, 2C9, and 2E1) did not contribute to monodeethylation of metoclopramide.

Formation of monodeethylmetoclopramide (pmol/min/pmol of P450) from metoclopramide (20 μM) by recombinant human P450 isoforms.
Data represent mean (n = 2 determinations). SeeMaterials and Methods for incubation conditions.
The formation of monodeethylmetoclopramide by recombinant human CYP2D6 followed simple Michaelis-Menten enzyme kinetics. The apparent kinetic parameters were derived using nonlinear regression analysis. The meanKm was 53.3 ± 4.1 μM, and the mean Vmax was 6.4 ± 4.5 pmol/min/pmol of P450 (n = 4) (Table 1). The data from recombinant human CYP2D6 are consistent with the data from HLMs and show a major involvement of CYP2D6 in the metabolism of metoclopramide. Because we noted an inhibitory effect of troleandomycin in HL09-14-99, we attempted to calculate kinetic parameters for monodeethylmetoclopramide formation by recombinant CYP3A4 as well (data not shown). Although the amount of metoclopramide metabolite formed increased with increasing concentrations of substrate by CYP3A4, we could not determine precise values due to unsaturable kinetics up to 200 μM substrate concentration.
Inhibition of P450 by Metoclopramide.
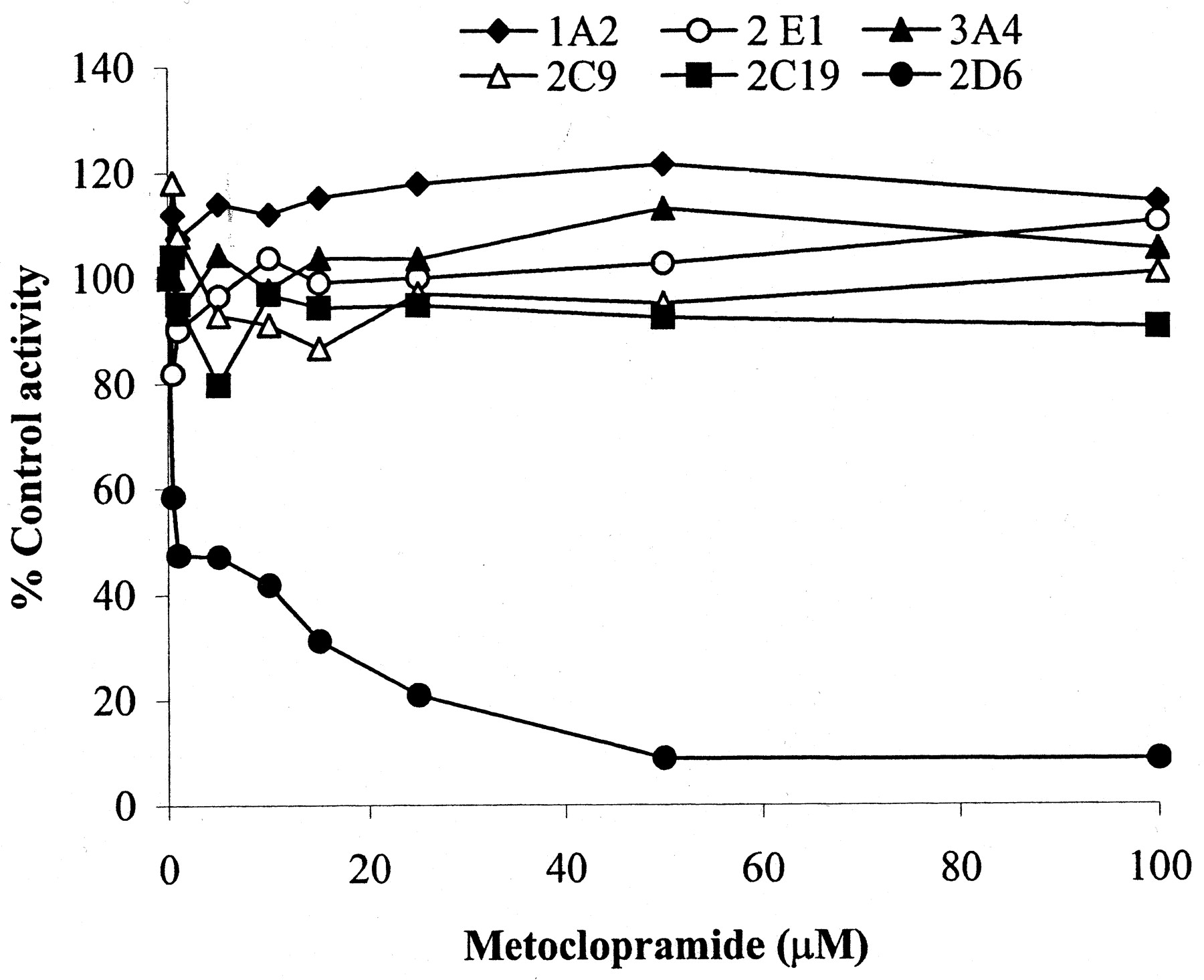
We investigated the ability of metoclopramide to inhibit P450 isoforms in HLMs through isoform-specific substrate probe reactions. A range of metoclopramide concentrations was incubated with a single concentration (around its observed Km in HLMs) of each isoform-specific substrate reaction probe. Metoclopramide was a potent inhibitor of CYP2D6 in HLMs (IC50, 1 μM), but the effect on other isoforms was negligible (Fig.6). As Fig.7 depicts, preincubation of metoclopramide (25 μM) with HLMs before the addition of dextromethorphan (25 μM) increased inhibition of dextrorphan formation (by 37 and 44% at 20- and 30-min preincubation times, respectively), suggesting the possibility of mechanism-based inactivation. This inactivation was dependent on the duration of incubation and inhibitor concentrations. The kinetic constants (the maximum inactivation at saturating metoclopramide,Kinact, and the concentration required to achieve the half-maximal rate of inactivation,K′i) describing the inactivation in microsomes at 37°C were determined by plotting the inverse of the rates of inactivation as a function of the inverse of the rates of inactivator concentration (K′i = 0.96 μM and Kinact = 0.026 min−1). A representative Dixon plot was constructed by incubating a range of metoclopramide concentrations (0–25 μM) and dextromethorphan (5–25 μM) in HLMs using the same preincubation protocol. The data in Fig.8 are consistent with potent competitive inhibition of CYP2D6-mediated dextromethorphanO-demethylation by metoclopramide, with a meanKi of 4.71 ± 1.33 μM (n = two different HLMs). Because there was no meaningful inhibition by metoclopramide of other P450 isoforms tested, we did not perform detailed experiments to estimate the inhibition constant of each isoform.

Inhibition of different P450 isoforms by metoclopramide (25 μM) in HLMs.
Used at concentrations approximating their respectiveKm values, isoform-specific substrate reactions probes were as follows: phenacetin (50 μM) for CYP1A2, flurbiprofen (5 μM) for CYP2C9, omeprazole (50 μM) for CYP2C19, dextromethorphan (25 μM) for CYP2D6, chlorzoxazone (25 μM) for CYP2E1, and midazolam (10 μM) for CYP3A4. Reaction velocities were expressed as a ratio relative to the velocity of controls (no inhibitor present). Each point represents duplicate measurements.

Effect of preincubation on the inhibition of different P450 isoforms by metoclopramide (25 μM).
Metoclopramide was preincubated with an NADPH-regenerating system and HLMs for 20 mins before addition of the specific substrate reaction probe. The effect of preincubation (%) in the controls and metoclopramide samples was calculated by dividing the velocity at each specific preincubation time to the velocity at 0 preincubation time. In the controls, no metoclopramide was used. Data represent measurement of duplicate incubates.
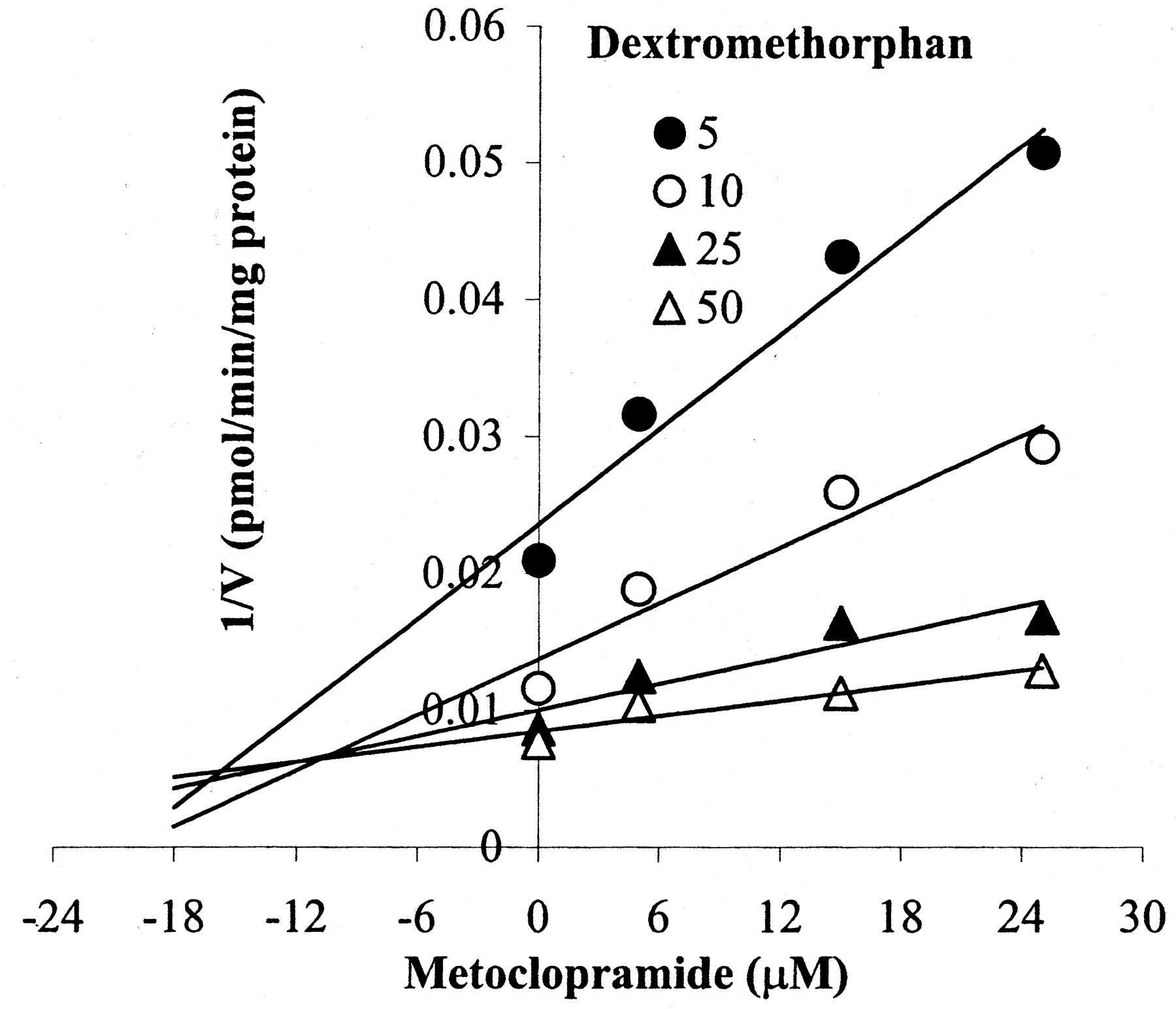
Dixon plot for the inhibition of CYP2D6 by metoclopramide (0–25 μM) in HLMs.
Metoclopramide was preincubated with HLMs for 20 min before addition of dextromethorphan (5–25 μM). Data are averages of duplicates.
Discussion
In this study, we present in vitro interaction of metoclopramide with the P450 system. We identified a new human metabolite of metoclopramide, monodeethylmetoclopramide, and found that CYP2D6 (and to some extent CYP3A) is the main isoform involved in its formation. We also demonstrated the ability of metoclopramide to inhibit drug-metabolizing P450 isoforms. These data should provide the basis not only for predicting drug interactions with metoclopramide but also to further evaluate the role of monodeethylation pathway in the action/toxicity of metoclopramide.
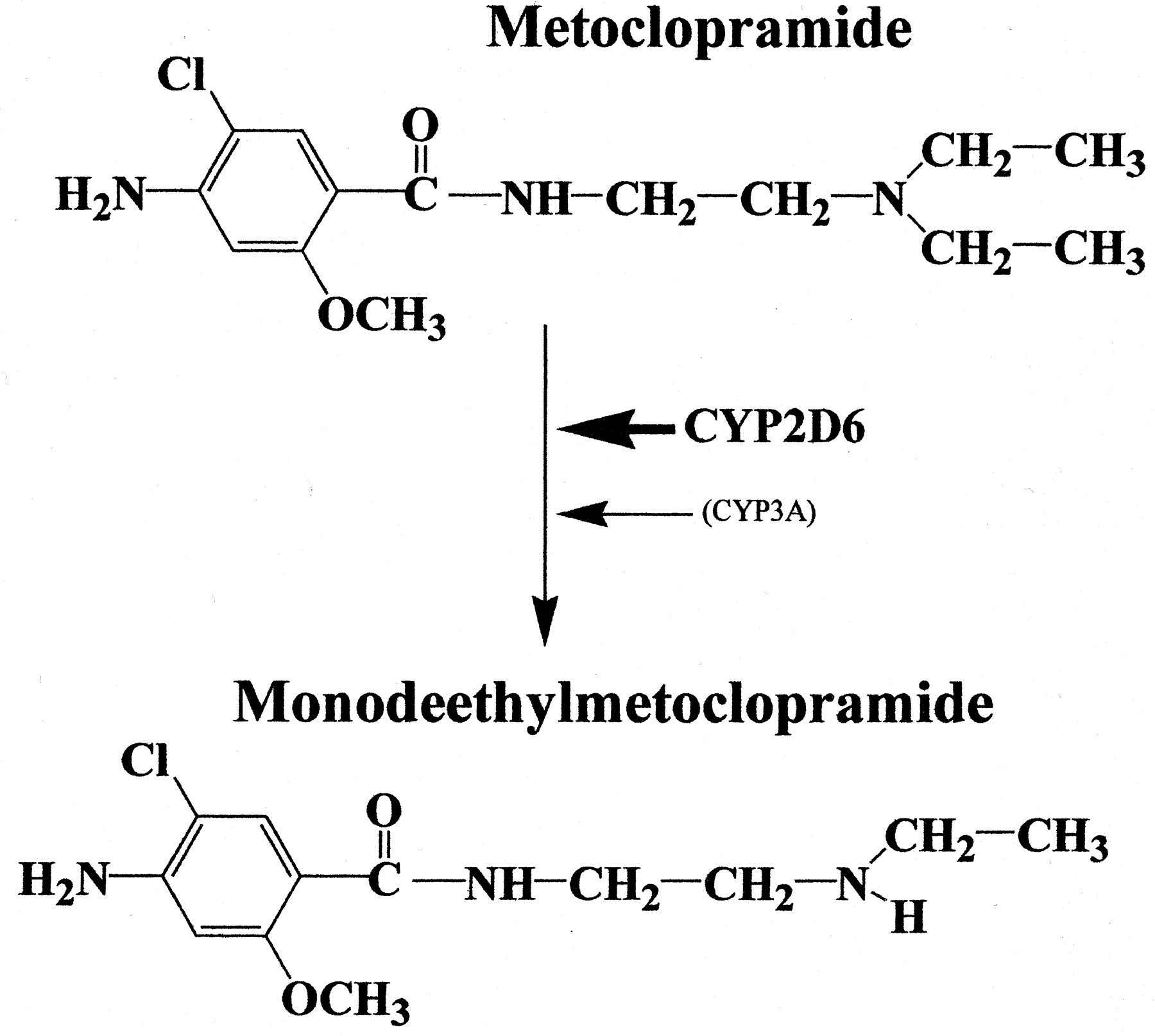
The human metabolism of metoclopramide has not been fully understood. Previous studies have shown that the major urinary metabolite of metoclopramide, which accounts for 30 to 40% of the dose, is theN-4-sulfate of metoclopramide (Bateman et al., 1980). A number of oxidative metabolites of metoclopramide have been identified in animals, but 2-[(4-amino-5-chloro-2-methoxybenzoyl)amino]acetic acid is the only metabolite reported in humans (Teng et al., 1977). According to Teng et al. (1977), monodeethylation is the primary metabolic pathway of metoclopramide in animals, but they failed to detect it in humans. Contrary to their findings, we have identified monodeethylmetoclopramide as a major oxidative metabolite of metoclopramide in in vitro HLMs (Fig. 9). The reason for the discrepancies between our data and those of Teng et al. (1977) is not clear. It is unlikely to be due to the lack of in vitro-in vivo correlation, but the metabolite analysis in urine (Teng et al., 1977) that involved complex procedures, including derivatization, might have hindered its proper identification in their samples.

Summary of metoclopramide metabolism in HLMs.
We characterized the isoforms involved in the monodeethylation of metoclopramide (Fig. 9). The data obtained strongly suggest that CYP2D6 is the main isoform. First, quinidine, a specific inhibitor of CYP2D6, was a potent inhibitor of metoclopramide monodeethylation in all HLMs tested (Ki < 3 μM). Second, the formation of monodeethylmetoclopramide was catalyzed by recombinant human CYP2D6 with the highest specific activity, and theKm values obtained from incubations with recombinant human CYP2D6 were consistent with those derived from HLMs. Third, metoclopramide was both a competitive and mechanism-based inhibitor of CYP2D6.
Do other P450 isoforms contribute to metoclopramide monodeethylation? CYP3A seems to contribute in some human livers but not in others. The fact that troleandomycin inhibited metoclopramide metabolism in HL09-14-99 (but not in HL161), that a significant residual activity was noted after quinidine, and that the combined effect of quinidine and troleandomycin almost completely abolished the formation of monodeethylmetoclopramide in this human liver support a role of CYP3A. In addition, recombinant human CYP3A4, although at relatively small specific activity compared with that of recombinant CYP2D6, has catalyzed metoclopramide monodeethylation. The small catalytic activity observed with other recombinant isoforms was not supported by our inhibition studies. Our data support the idea that CYP2D6 is the predominant isoform catalyzing metoclopramide deethylation with a contribution of CYP3A in some (but not all) HLMs. As shown in Fig. 5, metoclopramide metabolism was also catalyzed by recombinant human CYP1A2. However, given that furafylline (a specific inhibitor of CYP1A2) did not show any inhibition of metoclopramide metabolism in HLMs, it seems unlikely that CYP1A2 plays an important role.
Most CNS adverse effects of metoclopramide are concentration-dependent (Albibi and McCallum, 1983; Harrington et al., 1983). CYP2D6 is highly polymorphic, with 5 to 10% of the Caucasian population exhibiting poor metabolizer phenotypes (Bertilsson, 1995), and is inhibited by several drugs (Shin et al., 1999; Flockhart and Oesterheld, 2000). The contribution of CYP3A is likely to be significant when the alternative CYP2D6 metabolic pathway is blocked as a result of genetic polymorphism of CYP2D6 or coadministered drugs that inhibit this isoform, or in conditions where CYP3A is overexpressed. The activity of CYP3A is highly variable due to induction and inhibition by a large number of drugs, certain juices (e.g., grapefruit juice), and herbal drugs (e.g., St. John's wort), and as a result of genetic polymorphism in CYP3A5 (Kuehl et al., 2001). Since a significant amount of metoclopramide (up to 25% of the dose) is excreted unchanged via the urine (Harrington et al., 1983), the role of CYP2D6 (and CYP3A) is likely to be small and variable in patients whose renal function is normal. According toBateman et al. (1981), the clearance of a single dose of 10 mg of metoclopramide in renal failure patients was ∼30% of the normal patients and a 50% reduction of metoclopramide dose in these patients was recommended (Bateman et al., 1981). Patients with renal failure who also happen to have diminished CYP2D6 (and CYP3A) activity may even eliminate metoclopramide more slowly, causing an increase in plasma concentrations and adverse events. Consistent with this assumption, renal failure in combination with CYP2D6 poor metabolic status has been suggested to markedly increase the plasma concentrations of flecainide (Mikus et al., 1989; Evers et al., 1994), an antiarrhythmic that, like metoclopramide, is partly excreted by renal route unchanged and partly oxidized by CYP2D6. Monodeethylmetoclopramide has not been previously identified in humans, although it was a major urinary metabolite in rabbits, dogs, and rats (Teng et al., 1977). The formation of a toxic metabolite has been implicated in certain forms of metoclopramide-induced adverse effects such as dystonia and methemoglobinemia (Bateman et al., 1983). Whether the formation of this metabolite in vivo might contribute to the pharmacological actions of metoclopramide remains to be determined.
Metoclopramide is known to alter the pharmacokinetics of several coadministered drugs. Most of these effects could be understood in terms of the ability of metoclopramide to enhance gastric emptying and increased rate of absorption of these drugs, reflected by reduced time taken to achieve maximal plasma concentration (tmax) and increased maximal plasma concentration (Cmax) (Greiff and Rowbotham, 1994). However, there is not enough information about whether part of these effects might be mediated through inhibition by metoclopramide of P450-mediated drug metabolism. According to Dayer et al. (1992), metoclopramide (100 μM) was found to decrease the bioactivation (O-demethylation) of codeine to morphine, a reaction predominantly catalyzed by CYP2D6 (Sindrup et al., 1992), by ∼51%. In the present study, we investigated comprehensively the effect of metoclopramide on the most important drug-metabolizing enzymes, and we noted that metoclopramide was a potent inhibitor of CYP2D6 (meanKi values less than 5 μM) with no effect on other P450 isoforms. Additional inhibition of CYP2D6 by metoclopramide was observed during preincubation, suggesting that a mechanism-based inactivation may contribute to its overall inhibitory effect of metoclopramide. Since the Kivalues we documented here are within the therapeutic plasma concentrations after administration of the higher metoclopramide dose ends used for nausea and vomiting that occur during cancer chemotherapy [e.g., up to 0.6 to 4.5 μM at low- and high-dose therapy, respectively, and up to 6–13 μM at very high doses (Taylor and Bateman, 1983; Sallee et al., 2000)], reduced clearance of CYP2D6 substrate drugs should be expected during metoclopramide-concurrent administration. Particular attention should be given when a high dose of metoclopramide is coadministered with CYP2D6 substrates in critically ill patients and in patients with renal and hepatic failure, where metoclopramide appears to exhibit excessive accumulation (Bateman et al., 1981).
The number of patients who receive metoclopramide as a prokinetic is likely to increase in response to the recent withdrawal of cisapride from the market and because no new drug is yet available for this purpose, raising the potential of an increased incidence of adverse effects/interactions with this drug. Our in vitro findings show that metoclopramide undergoes monodeethylation primarily by CYP2D6 (and to some extent by CYP3A) and is a potent inhibitor of this isoform. We recognize that the significance of our in vitro results is yet to be confirmed in vivo through appropriate studies in humans. Until such studies are conducted, we recommend that physicians monitor patients for unexpected metoclopramide adverse effects, which may result from concurrently administered CYP2D6 inhibitor drugs (e.g., tricyclic antidepressants and neuroleptics) (Shin et al., 1999; Flockhart and Oesterheld, 2000) or genetic polymorphisms of CYP2D6 (and in some patients, of CYP3A5) and for the potential of slowed elimination of CYP2D6 substrates during metoclopramide administration.
Footnotes
-
This study was supported by the National Institute of General Medical Sciences Grants R01-GM56898-01 and T32-GM56898.
- Abbreviations used are::
- GI
- gastrointestinal
- P450
- cytochrome P450
- HLMs
- human liver microsomes
- HPLC
- high-performance liquid chromatography
- CNS
- central nervous system
- Received June 25, 2001.
- Accepted November 27, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}