


Abstract
The principal enzyme involved in the oxidation of mifepristone is cytochrome P450 3A4 (CYP3A4), which undergoes mechanism-based inactivation by the drug. However, no information is available on the interaction with CYP3A5, the second most abundant CYP3A enzyme in adult human liver. Oxidation of mifepristone by recombinant CYP3A4 produced mono- and didemethylated products and one C-hydroxylated metabolite, as reported previously. However, CYP3A5 produced only the demethylated metabolites. The apparent Vmax andKM values for formation of the monodemethylated product by CYP3A4 and CYP3A5 were 46 and 30 nmol/min/nmol P450, and 36 and 16 μM, respectively. Unlike CYP3A4, CYP3A5 was not inactivated by mifepristone. The basis of this differential susceptibility was explored using site-directed mutants in which a CYP3A4 residue was converted to its 3A5 counterpart. Surprisingly, none of these replacements caused a significant decrease in CYP3A4 inactivation by mifepristone. Examination of selected CYP3A4 mutants at 20 other positions indicated that the relative formation rate of the C-hydroxylated product could not account for the differential susceptibility of CYP3A4 and 3A5. Together these results indicate that mifepristone fails to orient itself in the CYP3A5 active site in such a way that its propylenic group is accessible for oxidation, thus rendering CYP3A5 unable to produce the C-hydroxylated product or putative ketene that leads to enzyme inactivation. Identification of mifepristone as a selective mechanism-based inactivation of CYP3A4 may be very useful in distinguishing between the two major CYP3A enzymes collectively responsible for the oxidative metabolism of over half of the drugs currently in use.
The antiprogestin mifepristone (RU 486), in combination with misoprostol, is the first clinically approved method of chemical abortion available for women in the US (DeHart and Morehead, 2001). The antiprogesterone activity of mifepristone has also shown promising potential against certain types of breast cancer, prostate cancer, meningioma, and uterine leiomyoma, as well as in the treatment of endometriosis (see review by Cadepond et al., 1997 and references therein). Mifepristone has also been reported to reverse P-glycoprotein-mediated drug resistance in vitro (Gruol et al., 1994; Lecureur et al., 1994), which could enhance its effectiveness as an anticancer agent alone or in combination therapy. Furthermore, the antiglucocorticoid activity of mifepristone also has several potential uses including the treatment of Cushing's syndrome, ectopic corticotropin secretion, adrenal carcinoma, and depression of human immunodeficiency virus replication (Cadepond et al., 1997).
The principal enzyme involved in the oxidation of mifepristone in human liver microsomes is cytochrome P450 3A4 (Jang et al., 1996), which is also inactivated by the drug (He et al., 1999). CYP3A4 and CYP3A5 are generally considered the two most important CYP3A enzymes in human liver. CYP3A5 is polymorphically expressed in 10 to 97% of adult livers and constitutes 6 to 100% of the entire hepatic CYP3A content (Wrighton et al., 1989, 1990; Hustert et al., 2001; Kuehl et al., 2001). These two CYP3A enzymes metabolize many of the same compounds and exhibit high (84%) amino acid sequence identity (Aoyama et al., 1989). However, compounds such as cyclosporin A (Aoyama et al., 1989), midazolam (Gorski et al., 1994), quinidine (Wrighton et al., 1990;Nielsen et al., 1999), and aflatoxin B1 (Wang et al., 1998) are known to show significant differences in their oxidation by CYP3A4 and CYP3A5. In contrast, to date not a single selective inhibitor/inactivator of CYP3A4 or CYP3A5 has been identified that could be used unequivocally to differentiate between these two major drug-metabolizing enzymes. Azole inhibitors such as ketoconazole and fluconazole, although showing slightly higher potency toward CYP3A4, also inhibit CYP3A5 (Gibbs et al., 1999). Triacetyloleandomycin, which forms a complex between the ferrous form of the heme iron and a nitroso intermediate, and the acetylenic steroid gestodene, which probably inactivates the enzyme via a ketene, also fail to cause selective inhibition of these two CYP3A enzymes (Wrighton et al., 1990; Chang et al., 1994). In the present study, we compare and contrast P450 inactivation and oxidation of mifepristone by recombinant CYP3A4 and CYP3A5. The study finds that mifepristone oxidation causes selective CYP3A4 inactivation. The structural basis of this differential susceptibility has been further explored using site-directed mutants.
Experimental Procedures
Materials.
Mifepristone, progesterone, 6β-hydroxyprogesterone, NADPH, CHAPS,1 and DOPC were purchased from Sigma-Aldrich (St. Louis, MO). Radiolabeled progesterone was obtained from PerkinElmer Life Sciences (Boston, MA). HEPES was obtained from Calbiochem Corp. (San Diego, CA). Oligonucleotide primers for PCR were either obtained from the University of Texas Medical Branch Molecular Biology Core Laboratory (Galveston, TX) or from Sigma-Genosys (The Woodlands, TX). The Expand PCR kit and Rapid Ligation kits were obtained from Roche Diagnostics (Indianapolis, IN). The QuikChange site-directed mutagenesis kit and GeneClean kit were from Stratagene (La Jolla, CA) and Qbiogene Inc. (Carlsbad, CA), respectively. Talon and Ni2+-metal affinity resin were from BD Biosciences Clontech (Palo Alto, CA) and Qiagen (Valencia, CA), respectively. Thin layer chromatography plates [silica gel, 250 μm, Si 250F (C19)] were purchased from J. T. Baker, Inc. (Phillipsburg, NJ). Recombinant NADPH-cytochrome P450 reductase and cytochrome b5 from rat liver were prepared as described earlier (Harlow and Halpert, 1997). All other chemicals were of the highest grade available and were obtained from standard commercial sources.
Construction of CYP3A4 Mutants.
Construction of mutants L210F and I369V was described previously (He et al., 1997; Wang et al., 1998). Mutants P107S, I120L, N206S, V376T, and L479T in the pCW expression vector and V296A in the pBS vector were reported by Wang et al. (1998). The V296A mutant was subcloned from pBS into the pSE380 expression vector using uniquePstI/StuI restriction sites. The mutant M371I was constructed by site-directed mutagenesis using the Expand High Fidelity PCR system (Roche Diagnostics). F108L, S478D, G480Q, and the multiple mutant R212K/D214G/F219L/T224I/V225L/I230T were generated using the QuikChange site-directed mutagenesis kit (Stratagene). To construct this multiple mutant, the R212K/D214G and F219L/T224I/V225L/I230T mutants were first generated, and the BamHI/PstI fragment containing the F219L//T224I/V225L/I230T mutation was subcloned into pSE3A4 R212K/D214G. The primers used for the construction of the various mutants are listed in Table 1. All constructs were sequenced to ensure that only the desired mutation was present (Protein Chemistry Core Laboratory, The University of Texas Medical Branch Galveston, TX).
Primers used for PCR
Expression and Purification of CYP3A4, CYP3A4 Mutants, and CYP3A5.
Wild-type CYP3A4 and most previously generated mutants were expressed as His-tagged proteins in Escherichia coli TOPP3 (or DH5α) cells and purified using Talon metal affinity resin (BD Biosciences Clontech) as described earlier (Domanski et al., 1998, 2001b; Khan et al., 2002, and references therein). The CYP3A4 → CYP3A5 mutants P107S, F108L, I120L, N206S, V296A, M371I, V376T, S478D, L479T, and G480Q, as well as the multiple mutant R212K/D214G/F219L/T224I/V225L/I230T were devoid of a His-tag and were assayed as preparations of CHAPS-solubilized membranes. Heterologous expression and purification of (His)6-tagged CYP3A5 using Ni2+-metal affinity chromatography (Qiagen) was performed as described previously (Domanski et al., 2001a;Hustert et al., 2001). The P450 contents were determined by carbon monoxide difference spectra in the presence of 1% Triton X-100. The Triton was added to the protein sample before dilution with 100 mM potassium phosphate, pH 7.3, 20% glycerol, 0.5% sodium cholate, 0.4% Renex, and 1.0 mM EDTA.
Mifepristone Oxidation Assay.
The reconstituted system contained 5 pmol of purified P450 enzyme, 10 pmol of rat liver cytochrome b5, 20 pmol of recombinant NADPH-cytochrome P450 reductase, 0.04% CHAPS, and 0.1 mg/ml DOPC. The mixture was incubated for 10 min at room temperature. A fixed concentration of mifepristone dissolved in 2% methanol (final) was added to the reconstituted protein system in 50 mM HEPES buffer, pH 7.6, and 15 mM MgCl2. The mixture was preincubated for 10 min at 37°C, and the reaction was initiated by adding NADPH (1 mM final). The total reaction volume of the assay was 100 μl. After 4 min of incubation at 37°C, the reactions were stopped by the addition of 200 μl acetonitrile. After centrifugation (3000 rpm, 5 min), 50 μl of the reaction mixture was analyzed by HPLC.
Separation of mifepristone metabolites was achieved at room temperature using an Ultrasphere ODS column (5 μm × 250 mm × 4.6 mm; Beckman Coulter, Inc., Fullerton, CA) with an Ultrasphere C18 guard column (5 μm × 7.5 mm × 4.6 mm; Alltech Associates Deerfield, IL). The HPLC system was described earlier (Khan and Halpert, 2000; Khan et al., 2002). After 5 min, the initial mobile phase of water/acetonitrile/methanol in a ratio of 55:25:20 (v/v/v) was changed over a period of 5 min to 25:40:35. The flow rate was 1.0 ml/min, and the UV detector was set at 304 nm.
LC-MS Analysis.
LC-MS analyses were performed on two different instruments. The initial LC-MS was performed at the School of Pharmacy, University of Arizona, Tucson, AZ. In this case, samples were prepared from incubations containing 100 pmol CYP3A4. The supernatant fractions were collected after centrifugation (3000 rpm, 10 min) and dried under a stream of nitrogen gas at room temperature. The dried sample was resuspended in the initial mobile phase before loading for LC-MS analysis. The LC-MS system consisted of a Finnigan-MAT LCQ instrument equipped with a Spectrasystem TSP P4000 HPLC system and an AS3000 auto sampler (Thermo Finnigan MAT, San Jose, CA). Automated acquisition of tandem mass spectometry spectra was carried out by data-dependent scanning with Finnigan Excalibur software. The total run time was set to 35 min. Metabolites were analyzed using the same Ultrasphere ODS C18 column (5 μm × 250 mm × 4.6 mm; Beckman Coulter, Inc.) used for HPLC analysis. The initial mobile phase composition of water/acetonitrile/methanol in ratios of 55:25:20 (v/v/v) was held for 5 min, then changed to 25:40:35 over the course of 15 min, and held for 10 min. A flow rate of 0.7 ml/min was achieved with a splitter tee upstream of a Microm Magic flow splitter box (Michrom BioResources Inc., Auburn, CA). The mass spectra were recorded under positive ion electrospray conditions, and the tandem mass spectometry spectra were obtained by collisional activation of MH+ parent ion species. The retention times of mifepristone and its three metabolites on the LC-MS instrument were slightly different from when using HPLC, because of different instrumentation and a small change in flow rate, but their elution order was the same on both instruments and as described in the literature (Heikinheimo et. al., 1990).
Later LC-MS analyses were carried out at the Core Facility, University of Texas Medical Branch Galveston, TX. Mass analysis was performed on a Micromass Q-Tof 2 mass spectrometer (Micromass UK Ltd., Manchester, UK) using an APCI ionization source operating in positive ion mode with an operating temperature of 350°C. HPLC separation was performed using a Waters CapLC capillary HPLC system (Waters, Corp., Milford, MA). The solvent flow rate was 5 μl/min with a gradient from 95:5 to 10:90 water/acetonitrile (v/v) over 75 min. Sample was introduced directly into the APCI source without flow splitting. Separation was achieved using a Waters symmetry C18 reversed phase column (0.3 mm × 150 mm). As expected, retention times of the metabolites in the two LC-MS instruments were different, but the fragmentation patterns were almost identical.
Inactivation Assays.
The inactivation assays were similar to those described previously (Khan et al., 2002). The preincubation mixture contained 35 pmol of purified P450, 70 pmol of rat liver cytochromeb5, 140 pmol of recombinant NADPH-cytochrome P450 reductase, 0.04% CHAPS, and 0.1 mg/ml DOPC. After incubation for 10 min at room temperature, mifepristone (dissolved in methanol, 1% final) was added to the reconstituted protein system in 50 mM HEPES buffer, pH 7.6, and 15 mM MgCl2. The protein-substrate mixture was further incubated for 2 min at 37°C, and the reaction was initiated by adding NADPH (1 mM final concentration). The total volume of the mixture was 560 μl. An aliquot (80 μl) of this mixture was taken at various time intervals (1–6 min), and added to a secondary reaction mixture (20 μl of volume) for determination of the residual progesterone hydroxylase activity ([progesterone] = 25 μM). The reaction was stopped after 6 min by the addition of 50 μl tetrahydrofuran and extracted with 1.0 ml of chloroform. The extracted solution (0.9 ml) was dried under nitrogen at 37°C, resuspended in 50 μl of chloroform, and spotted on the preadsorbent loading zone of a thin layer chromatography plate (Baker silica gel, 250 ml, Si 250F; J. T. Baker). The plate was developed twice in benzene:ethyl acetate:acetone (10:1:1, v/v/v). Metabolites of progesterone were visualized by autoradiography and identified by comparison with unlabeled standards. The radioactive areas from the plate were scraped into scintillation vials, and the metabolites were quantified by liquid scintillation counting.
Results
Mifepristone Oxidation and Inactivation of CYP3A4 and CYP3A5.
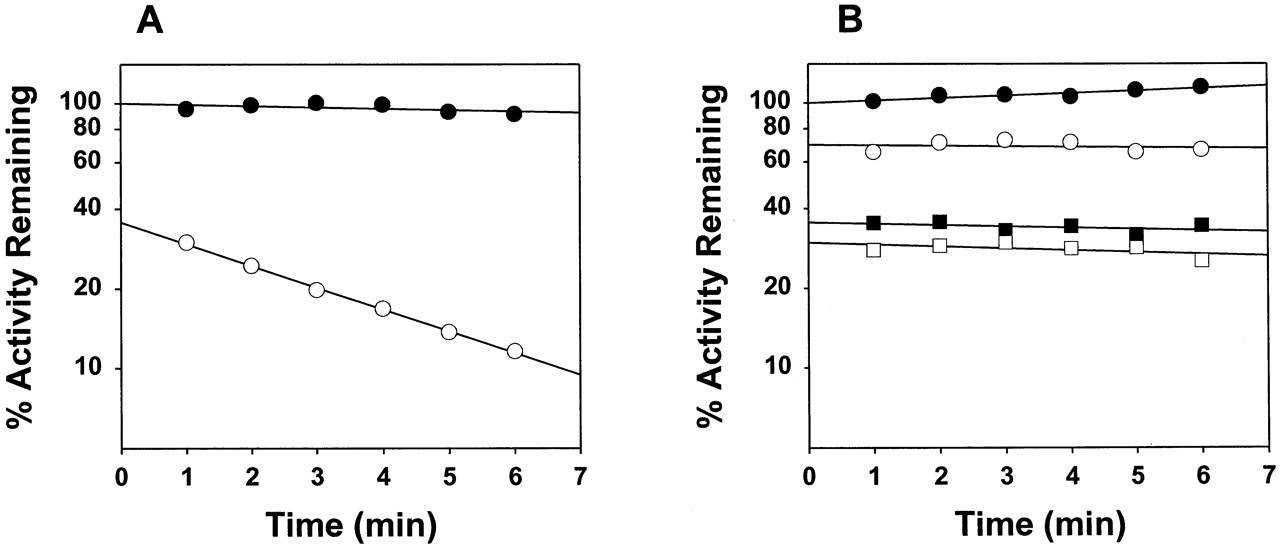
The time-course of mifepristone oxidation by CYP3A4 showed nonlinear behavior (data not shown), consistent with the inactivation of the enzyme (He et al., 1999). At 20 μM mifepristone, theki as measured by the decrease in the rate of progesterone 6β-hydroxylation was 0.17 min−1 (Fig. 1A, Table 2). The values forKI andkinactivation have been reported as 4.7 μM and 0.09 min−1, respectively (He, et al., 1999). In contrast to CYP3A4, CYP3A5 exhibited no time-dependent inhibition at mifepristone concentrations as high as 150 μM (Fig. 1B), although time-independent (presumably competitive) inhibition was clearly evident. Mifepristone at 20 μM also appears to be a more potent inhibitor of CYP3A4 than CYP3A5.

A, time-dependent inactivation of wild-type CYP3A4-dependent progesterone 6β-hydroxylase activity at 0 (●) and 20 (○) μM mifepristone; details are described under Experimental Procedures; the lines shown through experimental points were generated by linear regression analysis of the natural logarithm of the residual activity as a function of time; the rate constant of inactivation (k i) is derived from the negative slope of the line, whereas the extent of reversible inhibition is reflected by a decrease in the extrapolated activity at zero preincubation time compared with the methanol control; B, time- and concentration-dependent percentage decrease in progesterone 6β- hydroxylase activity of wild-type CYP3A5 at 0 (●), 20 (○), 100 (▪), and 150 (■) μM mifepristone.
Inactivation by mifepristone (20 μM) of wild-type and mutant CYP3A enzymes
CYP3A4-catalyzed mifepristone oxidation is known to produce at least three metabolites, one C-hydroxylated and two demethylated products (Heikinheimo et. al., 1990; Jang et al, 1996). HPLC analysis of mifepristone metabolism by recombinant CYP3A4 expressed in our laboratory also showed three metabolite peaks (data not shown). In the absence of standards, these metabolites were identified by LC-MS analysis (Fig. 2) The metabolite assignments were mainly based on the observation of mass at (m + 1)/z (where m signifies molecular weight of the metabolite) and the mass corresponding to loss of either the terminal propynyl group (in the case of mono- and didemethylated mifepristone) or of a hydroxylated propynyl group (in the case of C-hydroxylated mifepristone). The peaks at lower mass were almost identical for all three metabolites due to the presence of the same basic steroid skeleton. In contrast to CYP3A4, mifepristone oxidation by CYP3A5 showed formation of only the two demethylated products and no detectable C-hydroxylated product (data not shown).
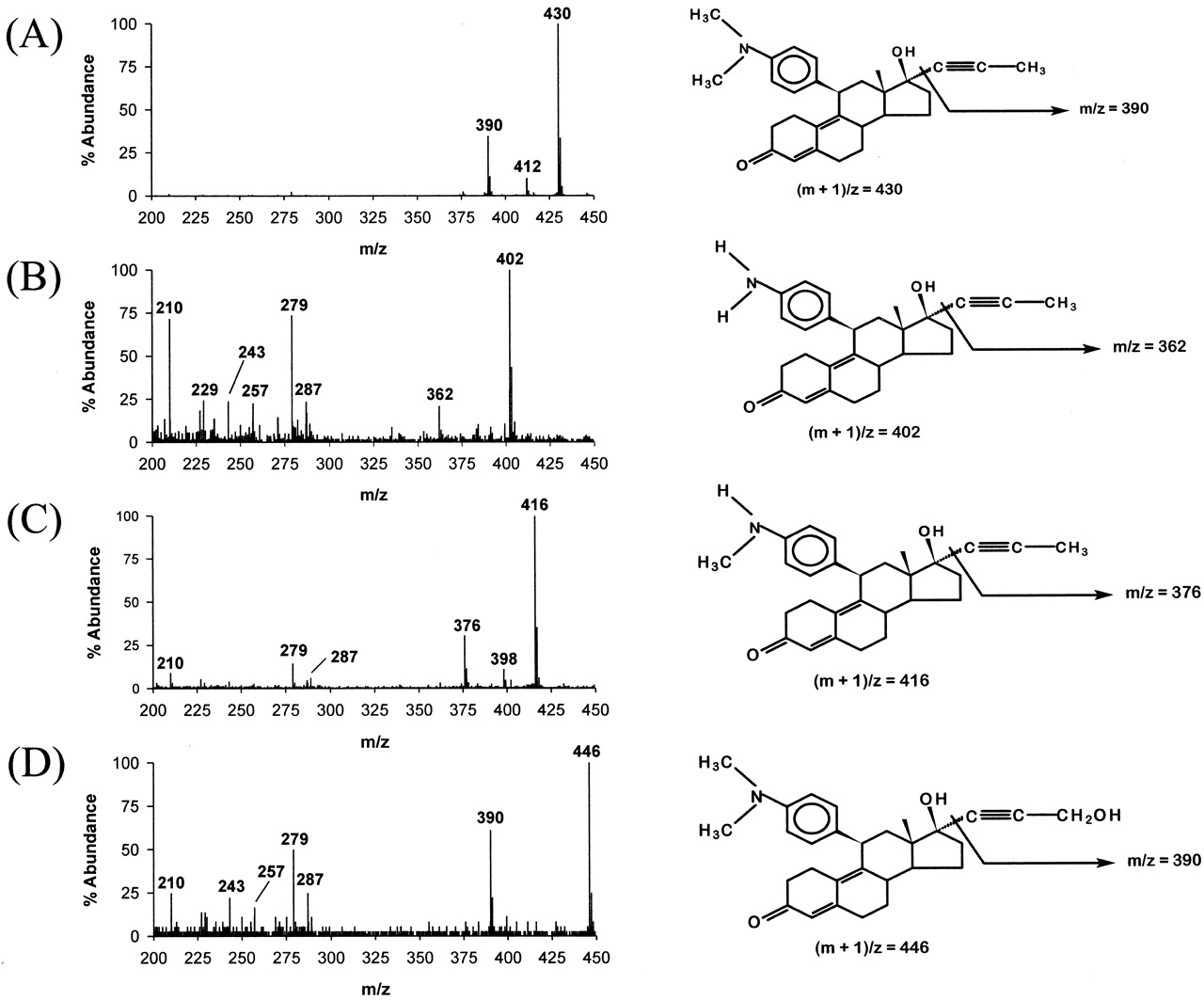
Mass spectra of (A) mifepristone and the three metabolites: (B) didemethylated mifepristone, (C) monodemethylated mifepristone, and (D) hydroxylated mifepristone, and their fragmentation patterns based on these peaks.
The steady-state kinetics of monodemethylated mifepristone formation by CYP3A4 were hyperbolic with Vmax andKM values of 46 ± 3 nmol/min/nmol P450 and 36 ± 5 μM, respectively (Fig.3). The steady-state kinetics of monodemethylated mifepristone formation by CYP3A5 were also hyperbolic with Vmax andKM values of 30 ± 1 nmol/min/nmol P450 and 16 ± 2 μM, respectively (Fig. 3).
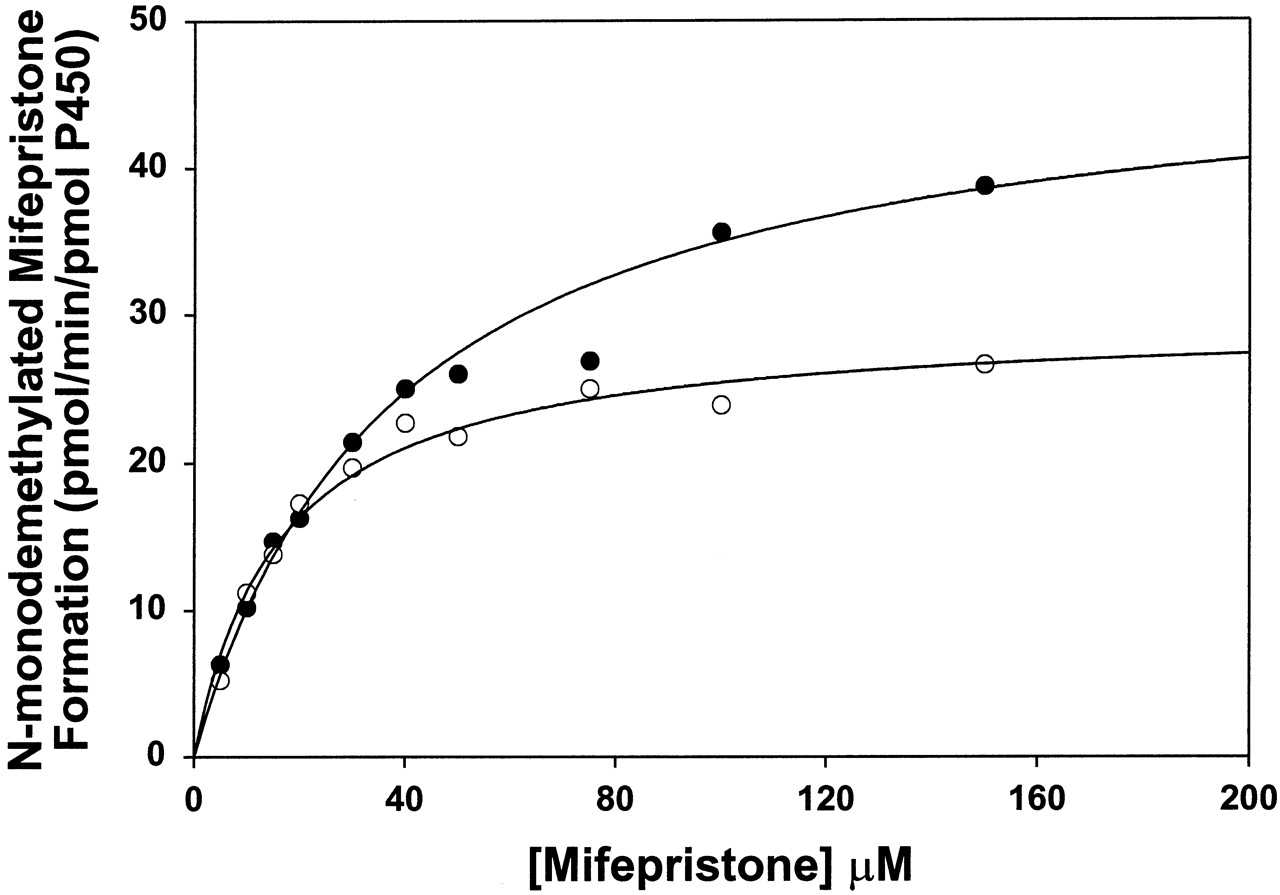
Steady-state kinetics of monodemethylated mifepristone formation by wild-type CYP3A4 (●) and CYP3A5 (○).
The lines through the experimental points show the fit to the Michaelis-Menten equation. Due to significant amount of substrate consumption at lower concentrations, the incubations were done using 5 pmol of the enzymes (see Experimental Procedures for details). Because of the low enzyme concentration used, the rates of didemethylated and hydroxylated mifepristone formation were too low to determine kinetic parameters for these metabolites.
Inactivation of CYP3A4 Site-Directed Mutants.
CYP3A4 and CYP3A5 differ by 78 of 503 amino acids, 12 of which fall within the 5 putative SRS domains (Aoyama et al., 1989, Wang et al., 1998; Domanski et al., 2001a) shown to be responsible for the substrate specificity of 3A4 with a wide range of compounds (Domanski and Halpert, 2001). Interestingly, none of the CYP3A4 → CYP3A5 mutants examined at these sites exhibited a significant decrease in susceptibility to inactivation by mifepristone (Table 2, bold). However, several of these residues have previously been reported as crucial for conversion of CYP3A4-catalyzed aflatoxin B1 oxidation to that of CYP3A5 (Wang et al., 1998; Xue et al., 2001). A multiple mutant R212K/D214G/F219L/T224I/V225L/I230T) that spans the proposed P450 substrate access channel (Poulos et al., 1986; Hasemann et al., 1995,Nakayama et al., 2001; Pikuleva et al., 2001) was also made to examine whether the substitution of these residues has a significant effect on CYP3A4 inactivation. However, this multiple mutant also showed mifepristone inactivation similar to that of the wild type (Table 2).
To investigate the differential susceptibility to mifepristone further, we also examined various other CYP3A4 SRS mutants. In initial experiments, more than 30 mutants at 20 SRS positions were analyzed for mifepristone oxidation by HPLC (data not shown). The residues studied included Thr-309, the counterpart of which has been proposed as crucial for proton delivery during oxygen cleavage by bacterial P450 enzymes (Hasemann et al., 1995; Poulos et al., 1995). Inactivation studies of P450 2B1, 2B4, and 2E1 by certain acetylenic compounds have shown an important role of this threonine residue (Roberts et al., 1996; Moreno et al., 2001 and references therein). Substitution of CYP3A4 Thr-309 with alanine or phenylalanine caused no significant effects on the rates of mifepristone oxidation or the metabolite profiles (data not shown), and the rate constant of inactivation was similar to that of the wild type (Table 2). The progesterone 6β-hydroxylation activity of T309F was too low to determine inactivation accurately.
Among the other individual SRS residues examined, A370F showed a metabolite profile very similar to that of the wild-type CYP3A5, whereas S119A showed the highest relative amount of C-hydroxylated mifepristone formation among all the mutants and wild-type enzymes examined (Fig. 4). The nonlinear time-dependence of mifepristone oxidation by CYP3A4 A370F and S119A suggested that their inactivation was similar to that of the wild type (data not shown). Inactivation experiments with the two mutants also revealed ki values similar to that of the wild type (Table 2).
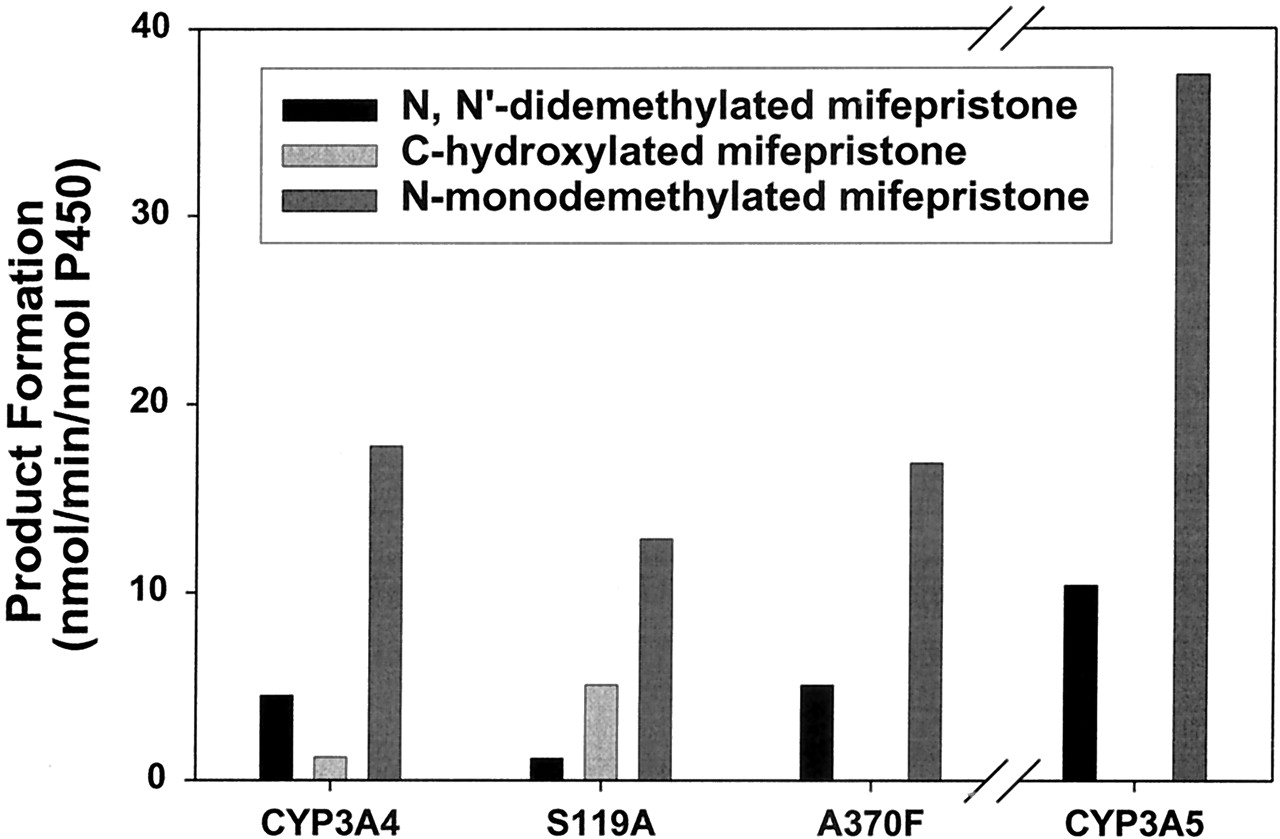
A comparison of the metabolic profiles of wild-type CYP3A4, CYP3A4 S119A, CYP3A4 A370F, and wild-type CYP3A5.
These reactions were performed using 25 pmol of enzyme and 100 μM mifepristone (see Experimental Procedures for details).
Discussion
CYP3A4 and 3A5 are the most important CYP3A enzymes found in adult human liver. Although 3A4 is generally considered the major CYP3A enzyme, a recent report suggests that the relative amount of 3A5 may be significantly higher in certain individuals than originally thought (Kuehl et al., 2001). Since CYP3A enzymes are involved in the oxidative metabolism of over half of the drugs available in the market, the variation in CYP3A content could be one of the most important contributors to interindividual and interracial differences in drug metabolism and disposition. However, the substrate specificities of CYP3A enzymes are well conserved within and across species, which has made it difficult to differentiate between these two human adult 3A forms. The absence of a suitable genetic marker has further complicated the process of assessing the contribution of these two enzymes in individuals (Hustert et al., 2001). The identification of mifepristone as a selective inactivator of CYP3A4 should provide an excellent probe for differentiating between the two major human CYP3A enzymes.
The mechanism-based inactivation of CYP3A4 by mifepristone has been ascribed to protein rather than heme modification (He et al., 1999). By analogy with compounds bearing a terminal acetylenic or propylenic group (Ortiz de Montellano and Kunze, 1980; Foroozesh et al., 1997;Roberts et al., 1997), it has been proposed that mifepristone is oxidized and converted via a 1,2-methyl shift to a ketene, which subsequently reacts with one or more nucleophiles present in the active site of CYP3A4. A series of earlier studies involving the isolation and characterization of the radiolabeled peptide containing the covalently attached inactivator and the use of site-directed mutagenesis implicated a conserved threonine residue in the I-helix of P450 2B1, 2B4, and 2E1 in the inactivation of these enzymes by various compounds (Roberts et al., 1993, 1994, 1996; Moreno et al., 2001). However, substitution of the corresponding residue (Thr-309) in CYP3A4 by alanine showed no significant effect on enzyme inactivation by mifepristone. Previous mutagenesis studies from our laboratory also have indicated that conservation of this threonine residue is not essential for substrate metabolism by CYP3A4 (Domanski et al., 1998,2001b; Khan et al., 2002). The unaltered susceptibility to mifepristone inactivation of various CYP3A4 → CYP3A5 mutants was rather surprising, since several of these substitutions (N206S and L210F) are crucial for conversion of CYP3A4 to CYP3A5 regioselectivity with aflatoxin B1 and testosterone (Wang et al., 1998;Xue et al., 2001). A recent study by Lightning et al. (2000) has implicated residue Glu-307 in the mechanism-based inactivation of CYP3A4 by L-754,394 based on molecular modeling. However, CYP3A4 and CYP3A5 contain the same amino acid at this position, so it is less likely that this residue may be involved in mifepristone selectivity. Thus, the identity of the residue(s) that leads to the differential susceptibility of CYP3A4 and CYP3A5 to mifepristone remains elusive. It is possible that CYP3A4 inactivation by mifepristone involves more than one amino acid or a residue not explored in this study.
The other major difference between mifepristone oxidation by CYP3A4 and CYP3A5 is the absence of C-hydroxylated product formation in the case CYP3A5 (Fig. 4). Since formation of this product and of the ketene intermediate proposed to lead to CYP3A4 inactivation involves the same propylenic group, the results suggest that mifepristone is not able to orient itself in the CYP3A5 active site in such a way that the propylenic group is accessible for oxidation. In contrast, the inactivation of CYP3A4 by mifepristone is likely to depend upon the factors that influence the partitioning between the oxidation of the terminal methyl group and formation of the putative ketene intermediate.2 In the case of CYP3A4 A370F, which gets inactivated but does not produce the C-hydroxylated product, substitution of the active site residue appears to favor only formation of the putative ketene intermediate and not the terminal alcohol.
An earlier study indicated that altered pharmacokinetics and metabolism of mifepristone have no role in its ineffectiveness in the termination of pregnancy in 10–15% of women (Heikinheimo et al., 1990). Therefore, the differences in the metabolism of mifepristone by CYP3A4 and CYP3A5 may not alter its efficacy as an abortifacient. However, the use of mifepristone for the treatment of other diseases may need to be further evaluated in light of our findings. In addition, selective inactivation of CYP3A4 but not CYP3A5 by mifepristone could significantly affect the bioavailability and clearance of various coadministered drugs that are metabolized by these two CYP3A enzymes. Drugs such as midazolam, quinidine, and cyclosporin A, which are differentially metabolized by CYP3A4 and CYP3A5, may be of greatest concern. Similarly, the relative risk upon exposure to the heptocarcinogen aflatoxin B1 may be significantly enhanced by this selective enzyme inactivation, because oxidation by CYP3A5 largely produces the genotoxic exo-8, 9-epoxide, as opposed to the innocuous 3α-hydroxylated product predominantly formed by CYP3A4. A recent report suggesting that CYP3A5 is more frequently expressed in livers of African Americans than those of Caucasians (Kuehl et al., 2001) further highlights the importance of proper evaluation of the selective contribution of these two enzymes in individuals to avoid adverse drug reactions.
Acknowledgments
We thank Dr. Tammy L. Domanski, Dr. Fabienne Roussel, You Ai He, E. Licad-Coles, and Ryan Dick for providing the mutants; Dr. Anthony Haag (Mass Spectrometry Laboratory) for his help with LC-MS analysis; and Dr. Emily Scott for critical reading of the manuscript.
Footnotes
-
This work was supported by Grants GM54995 (to J.R.H.), DK26506 (to M.A.C.), and Core Center Grant ES06676 from the National Institutes of Health.
-
↵2 Nonenzymatic hydrolysis of ketene intermediates is known to produce the corresponding acid derivatives. Our attempts to isolate and/or identify the mifepristone acid derivative by HPLC and LC-MS were unsuccessful. These experiments were performed with wild-type CYP3A4 as well as two mutants, S119A and A370F, which form the maximal and almost negligible amount of C-hydroxylated mifepristone, respectively.
- Abbreviations used are::
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammino]-1-propanesulfonic acid
- DOPC
- dioleoylphosphatidylcholine
- PCR
- polymerase chain reaction
- HPLC
- high-performance chromatography
- LC-MS
- liquid chromatography-mass spectometry
- APCI
- atmospheric pressure chemical ionization
- SRS
- substrate recognition site
- Received April 12, 2002.
- Accepted June 3, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}