


Abstract
Pregnane X receptor (PXR) and constitutive androstane receptor (CAR) are key regulators of xenobiotic-inducible cytochrome P450 gene expression. Whereas much is known about their role in regulating drug metabolism, little is known regarding their role in regulating drug transport in vivo. Wild-type mice and mice lacking PXR (PXR-KO) were used to examine the inducible expression of two drug transporter genes, Oatp2 (Slc21a5) andMrp3 (Abcc3), in liver following treatment with selective PXR and CAR activators. Selective activation of PXR or CAR induced Oatp2 and Mrp3expression in wild-type mice but not in PXR-KO mice. Basal expression levels of Oatp2 and Mrp3 gene were significantly higher in PXR-KO mice when compared with wild-type mice. Additionally, phenobarbital (PB)-inducible Oatp2 andMrp3 gene expression was significantly increased in the PXR-KO mice when compared with wild-type PB-treated mice. We also examined the effect of PXR ablation on PB-inducible hepatic CYP3A activity in vivo. Microsomes isolated from PB-treated PXR-KO mice exhibited a significantly elevated rate of testosterone 6β-hydroxylation when compared with microsomes isolated from wild-type PB-treated mice. PB treatment produced significantly increased levels of hepatomegaly in PXR-KO mice when compared with wild-type PB-treated mice. Taken together, these results suggest that nonliganded PXR plays a net negative role in coregulating shared PXR/CAR-target gene expression in vivo and extend the hypothesis that PXR and CAR coregulate not only drug metabolism but also drug transport.
Cytochromes P450 (P450s1) are a superfamily of heme-thiolate-containing proteins involved in the oxidative metabolism of steroid hormones, bile acids, fatty acids, and prostaglandins (Nelson et al., 1996). Additionally, a wide range of compounds including carcinogens, environmental pollutants, and drugs are metabolized by P450s (Maurel, 1996). The net effect of such metabolism converts parent molecules into suitable substrates for drug transporter proteins in liver, kidney, and intestine. Thus, drug metabolism and drug transport function coordinately to prevent the accumulation of toxic chemical compounds.
We have shown that pregnane X receptor (PXR, NR1I2) activation mediates the inducible expression of CYP3A in mice (Kliewer et al., 1998; Staudinger et al., 2001a,b). Biochemical studies suggest that PXR and CAR bind the same or overlapping enhancer elements within the promoters of CYP3A and CYP2B genes in liver cells in a competitive manner (Xie et al., 2000; Goodwin et al., 2001, 2002;Smirlis et al., 2001). Although much is known about the role these two receptors play in coregulating P450 gene expression, comparatively little is known about their role in coregulating drug transporter gene expression in vivo.
To determine whether drug transporter gene expression is coregulated by PXR and CAR activation in vivo, we examined the expression levels of organic anion-transporting polypeptide (Oatp2,Slc21a5) and multi-drug resistance associated protein 3 (Mrp3, Abcc3) following treatment with selective activators of PXR and CAR in wild-type mice and mice lacking PXR (PXR-KO). Although biochemical experiments show that PXR/CAR crosstalk likely plays a role in coregulating P450 gene expression, to our knowledge, no data exist describing the biological consequence of PXR/CAR crosstalk in vivo. To examine the biological consequence of PXR/CAR crosstalk at the level of P450 activity in vivo, we determined the rate of testosterone 6β-hydroxylation in hepatic microsomes isolated from wild-type and PXR-KO mice following selective activation of PXR or CAR. Our data reveal that selective activation of PXR and CAR regulates the inducible expression of the drug transporter genesOatp2 and Mrp3. In addition, our data suggest that nonliganded PXR plays a negative or competitive pharmacological role in regulating drug metabolism and drug transport in mice.
Materials and Methods
Maintenance and Treatment of PXR-KO and Wild-Type Mouse Populations.
Generation of the PXR-KO mice was previously described (Staudinger et al., 2001b). Adult male wild-type mice and PXR-KO mice were maintained on standard laboratory chow and were allowed food and water ad libitum. All mice were treated once a day i.p. with vehicle (corn oil, saline), pregnenalone 16α-carbonitrile (PCN) (400 mg/kg in corn oil), PB (100 mg/kg in saline), or 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) (3 mg/kg in corn oil) for 4 days.
RNA Isolation and Northern Blot Analysis.
Total RNA was isolated from liver using a commercially available reagent (Trizol; Invitrogen. Carlsbad, CA) according to the manufacturer's instructions. Total RNA (10 μg) was resolved on a 1% agarose/2.2 M formaldehyde denaturing gel and transferred to a nylon membrane (Hybond N+; Amersham Biosciences Inc., Piscataway, NJ). Blots were hybridized with 32P-labeled cDNAs corresponding to sequences for Oatp2 (bases 1–240, Genbank number AB031814), Mrp3 (bases 1705 to 2136, GenBank NW_000040), Cyp3a11 (bases 69 to 1609, GenBank NM 007818), and β-actin (BD Biosciences Clontech, Palo Alto, CA).
Real-Time PCR Analysis.
Total RNA was isolated from liver as described and 1 μg was reverse-transcribed in a 20-μl volume using random primers as described by the manufacturer (Promega, Madison, WI). Equal amounts of reverse-transcribed cDNA were used in real-time quantitative polymerase chain reaction (rtQ-PCR) reactions in the Cepheid Smart Cycler (Sunnyvale, CA) and included 200 nM fluorogenic probe and 150 nM primers specific for Oatp2, Mrp3,Cyp3a11, or β-actin (Table1). Cycling conditions were 95°C for 2 min followed by 45 cycles of 95°C for 15 s, 62°C for 15 s, and 72°C for 15 s. Fold-induction was calculated as described (Schmittgen et al., 2000), and the data were normalized using β-actin as an internal standard. The BLASTN function at the National Center for Biotechnology Information website was used to assess the specificity of all primers and fluorogenic probes.
Primer-probe sets
Relative Liver Weight.
Five mice were randomly allocated per treatment group. Mice were weighed both before and after 4 days of treatment. Livers from mice pretreated with vehicle, PCN, or PB were removed and weighed on the morning of day five following 4 days of treatment. The data are expressed as grams of liver per 100 g of body weight.
Preparation of Microsomes and HPLC Testosterone 6β-Hydroxylation Assay.
Liver microsomes were prepared as previously described (Pearce et al., 1996). Protein concentration of isolated microsomal preparations was determined with the bicinchoninic acid Protein Assay Reagent kit (Pierce Chemical, Rockford, IL) as described by the manufacturer. Microsomal testosterone 6β-hydroxylase activities were determined as described previously (Pearce et al., 1996). The data are expressed as picomoles per minute per milligram of protein.
Statistical Analyisis.
Differences between liver mass, messenger RNA levels, and enzymatic activities in vehicle, PCN-, PB-, and TCPOBOP- treated animals were determined using a one-way analysis of variance followed by the Duncan's multiple range post hoc test.
Results and Discussion
PXR and CAR Activation Induces Oatp2 andMrp3 Gene Expression in Vivo.
We qualitatively examined Oatp2, Mrp3, andCyp3a11 gene expression in wild-type mice following treatment with PCN, PB or TCPOBOP using total RNA isolated from liver in Northern blot analysis. The cDNA probes used in Northern analysis for Oatp2 and Mrp3 were generated against unique sequences and are specific (see Materials and Methods). Use of the BLAST sequence analysis program revealed that there are six mouse CYP3A family members. The cDNA probe forCyp3a11 Northern analysis spans regions that have high homology. Thus, while the Oatp2 and Mrp3 Northern probes are specific, we feel it likely that we are visualizing changes in expression of multiple mouse CYP3A family members in our Northern analysis.
Treatment of wild-type mice with the PXR-selective activator PCN produced a large increase in Oatp2, Mrp3, andCYP3A gene expression levels (Fig.1A, lanes 4–6 versus lanes 1–3). Treatment of wild-type mice with the CAR-selective activator PB produced only slight increases in Oatp2, Mrp3, and CYP3A gene expression levels (Fig. 1A, lanes 7–9 versus lanes 1–3). In contrast to treatment with PB, treatment of wild-type mice with the CAR-selective activator TCPOBOP produced large increases in Oatp2, Mrp3, and CYP3A gene expression levels (Fig. 1A, lanes 10–12 versus lanes 1–3). Identical Northern blot analysis of total liver RNA isolated from PXR-KO mice reveals that PCN produced no change in Oatp2, Mrp3, andCYP3A gene expression relative to PXR-KO vehicle-treated controls (Fig. 1B, lanes 4–6 versus lanes 1–3). Strikingly, PB treatment of PXR-KO mice produced relatively large increases inOatp2, Mrp3, and CYP3A gene expression when compared with the PB-treated wild-type mice (Fig. 1B, lanes 7–9 versus Fig. 1A, lanes 7–9). Treatment of PXR-KO mice with TCPOBOP produced modest increases in the expression of these three genes when compared with the vehicle-treated PXR-KO mice (Fig. 1B, lanes 10–12 versus Fig. 1B, lanes 1–3).
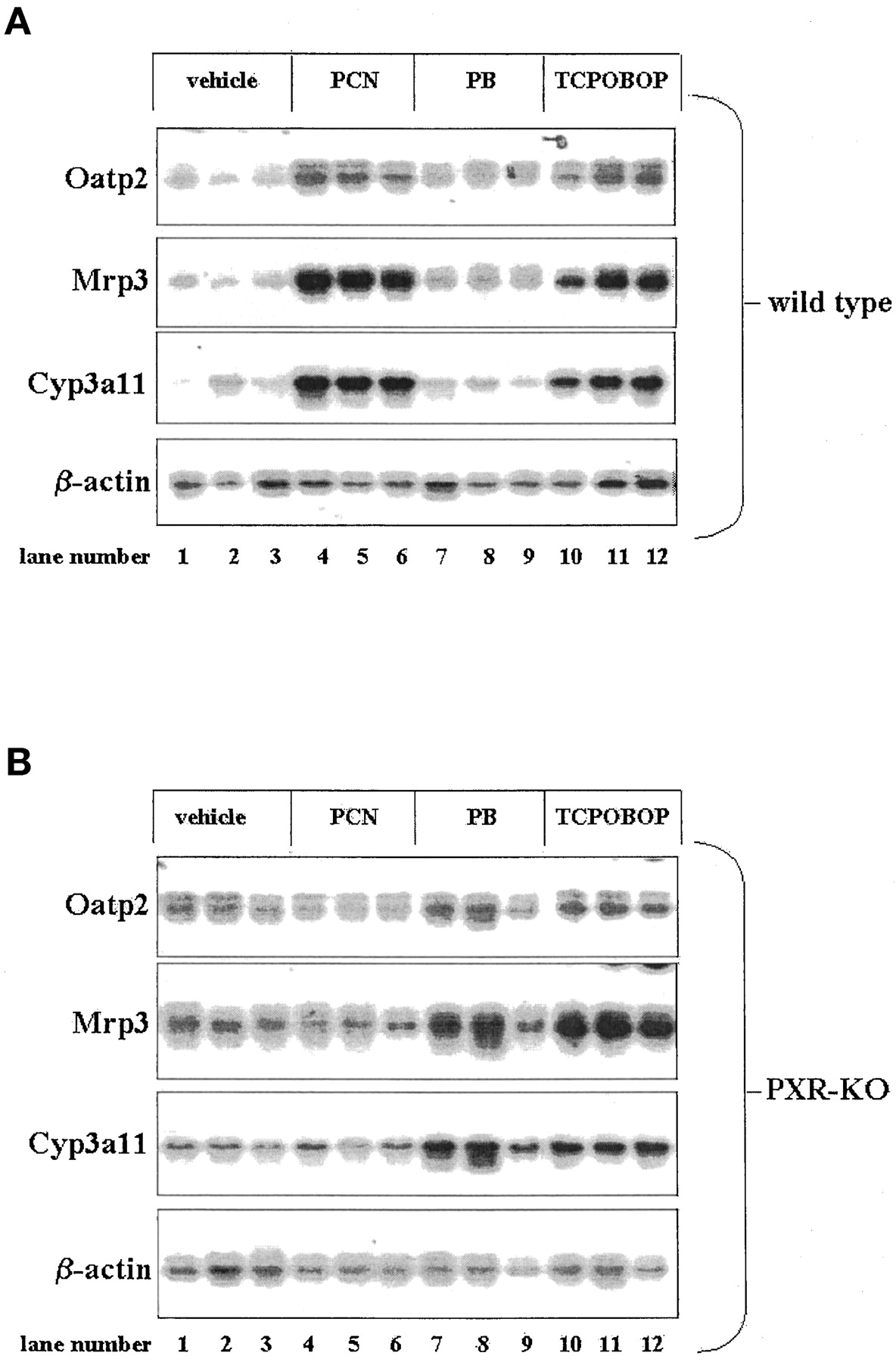
Induction of Oatp2, Mrp3, and Cyp3a11 expression by PCN, PB, and TCPOBOP in wild-type and PXR-KO mice.
Three wild-type (A) and PXR-KO (B) mice per experimental group were treated for 4 days i.p. with vehicle, PCN (400 mg/kg), PB (100 mg/kg), or TCPOBOP (3 mg/kg). On the morning of day five, total liver RNA was isolated and run on a 1% agarose gel. After transferring to nylon membrane the blot was probed, exposed, stripped, and subsequently re-probed with 32P-labeled cDNA probes for mouseOatp2, Mrp3, Cyp3a11, and β-actin.
To quantify relative expression levels of Oatp2,Mrp3, and Cyp3a11 in wild-type and PXR-KO mice, we developed rtQ-PCR probe sets specific for these three genes. The probe sets were designed to account for the fact that all three genes belong to gene families and were generated against unique lengths of cDNA sequence (Table 1). DNA sequence and BLAST analysis of each probe set and amplimer confirmed the specificity of our analysis (data not shown).
We first examined hepatic Oatp2 gene expression in wild-type and PXR-KO mice (Fig. 2A). A comparison of vehicle-treated mice revealed that the basal expression ofOatp2 was 2.4 ± 0.3-fold higher in the PXR-KO mice. Treatment with PCN induced expression of Oatp2 by 10.1 ± 1.4-fold in wild-type mice but had no significant effect onOatp2 expression PXR-KO mice when compared with vehicle-treated PXR-KO mice. Treatment of wild-type mice with PB did not induce Oatp2 expression significantly (1.2 ± 0.1-fold). In contrast, treatment of PXR-KO mice with PB produced robust induction of Oatp2 expression (9.6 ± 1.6-fold). Treatment with TCPOBOP induced Oatp2 expression in both wild-type (8.9 ± 0.9-fold) and PXR-KO (6.2 ± 0.3-fold) mice, respectively, when compared with vehicle-treated wild-type mice.

Quantification of Oatp2, Mrp3, and Cyp3a11 gene expression levels.
RNA from three individual wild-type or PXR-KO mice per experimental group (vehicle, PCN, PB, and TCPOBOP) was pooled, and rtQ-PCR analysis was performed using primers and probes specific forOatp2 (A), Mrp3 (B), andCyp3a11 (C), and β-actin as described underMaterials and Methods. The data are expressed as -fold induction relative to the wild-type vehicle experimental group and are reported as the mean ± S.E.M. of three independent experiments. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05).
We next examined hepatic Mrp3 gene expression in wild-type and PXR-KO mice (Fig. 2B). Similar to Oatp2, basalMrp3 gene expression was 2.0 ± 0.4-fold higher in PXR-KO mice. Treatment with PCN induced expression ofMrp3 by 5.9 ± 0.5-fold in wild-type mice but had no significant effect on Mrp3 expression levels in PXR-KO mice. Interestingly, treatment of wild-type mice with PB did not induce Mrp3 expression significantly (1.6 ± 0.2-fold), whereas treatment of PXR-KO mice with PB produced strong induction of Mrp3 expression (6.0 ± 0.9-fold). As observed with Oatp2, treatment with TCPOBOP inducedMrp3 expression in both wild-type (5.9 ± 0.5-fold) and PXR-KO (6.0 ± 0.9-fold) mice, respectively.
Because the CYP3A family represents the prototypical PXR/CAR-shared target gene, we examined hepatic expression ofCyp3a11 in wild-type and PXR-KO mice (Fig. 2C). Treatment with PCN induced expression of Cyp3a11 19.1 ± 1.1-fold in wild-type mice but had no effect on Cyp3a11 expression levels in PXR-KO mice. Remarkably, treatment of wild-type mice with PB produced modest induction of Cyp3a11 expression (8.9 ± 2.1-fold), whereas treatment of PXR-KO mice with PB produced strong induction of Cyp3a11 expression (23.8 ± 1.1 fold). Treatment with TCPOBOP induced Cyp3a11 expression in both wild-type (19.0 ± 1.9 fold) and PXR-KO (18.5 ± 1.9 fold) mice, respectively.
Nonliganded PXR Inhibits PB-Inducible Testosterone 6β-Hydroxylation and PB-Mediated Hepatomegaly in Vivo.
Because PXR-KO mice exhibited higher basal expression levels ofOatp2, Mrp3, and Cyp3a11, we examined the rate of testosterone 6β-hydroxylation in hepatic microsomes isolated from the livers of vehicle-treated wild-type and PXR-KO mice (Fig. 3A). The basal rate of testosterone 6β-hydroxylation was approximately 3-fold higher in the vehicle-treated PXR-KO mice when compared with vehicle-treated wild-type mice (2060 ± 200 pmol/min/mg versus 685 ± 150 pmol/min/mg). In wild-type mice, PCN-treatment produced a 17-fold increase in the rate of microsomal testosterone 6β-hydroxylation whereas PB treatment produced a 4.6-fold increase. Treatment of PXR-KO mice with PCN had no effect on the rate of microsomal testosterone 6β-hydroxylation. Surprisingly, the overall rate of testosterone 6β-hydroxylation was approximately 227% higher in the PB-treated PXR-KO mice when compared with PB-treated wild-type mice (10,300 ± 900 pmol/min/mg versus 3160 ± 300 pmol/min/mg), suggesting that nonliganded PXR plays a negative role in regulating the enzymatic activity of CYP3A family members in mice.

Induction of testosterone 6β-hydroxylation and hepatomegaly caused by PCN and PB.
A, wild-type and PXR-KO mice were treated for 4 days i.p. with vehicle, PCN (400 mg/kg), or PB (100 mg/kg). Livers were collected on the morning of day five, and microsomal preparations were isolated as described under Materials and Methods. The data are expressed as the mean ± S.E.M. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). B, mice were treated with vehicle (corn oil or saline), PCN (400 mg/kg), or PB (100 mg/kg) for four days. On the morning of the fifth day, the body weight and the liver weight were measured. The percentage of liver weight relative to total body weight is represented as mean ± S.E.M. and was determined in at least five animals. Letters different from each other indicate a statistical difference between treatment groups (p< 0.05).
Acute treatment of mice with PB increases relative liver weight in a CAR-dependent manner and is thought to be a reflection of increases in cellular hypertrophy and hyperplasia (Wei et al., 2000). We therefore examined the relative liver weights of wild-type and PXR-KO mice following treatment with the selective PXR and CAR activators PCN and PB (Fig. 3B). Treatment of wild-type mice with PCN or PB produced a significant increase in relative liver weight; with PCN producing a 66% increase (from 3.9 ± 0.3 to 6.5 ± 0.3) and PB treatment producing a 33% increase (from 3.9 ± 0.3 to 5.2 ± 0.2). PCN treatment of PXR-KO mice produced no increase in relative liver weight when compared with the vehicle-treated PXR-KO group. Treatment of PXR-KO mice with PB produced a 74% increase in relative liver weight when compared with the vehicle-treated PXR-KO mice (from 3.8 ± 0.2 to 6.6 ± 0.3). Strikingly, mice that lack PXR exhibit a 27% greater increase in relative liver weight in response to PB treatment when compared with the PB-treated wild-type mice (6.6 ± 0.3 in PB-treated PXR-KO mice versus 5.2 ± 0.2 in PB-treated wild-type mice).
Our results demonstrate that shared PXR/CAR-target genes are more efficaciously induced by PB treatment in mice that lack PXR. It is interesting to note that this effect is absent when CAR is activated using TCPOBOP. One potential reason for this discrepancy could be that higher basal expression of PXR/CAR-target genes in PXR-KO mice has differential effects upon the disposition of PB and TCPOBOP in vivo. Alternatively, these data may reflect the fact that PB activates CAR through an indirect mechanism, whereas TCPOBOP serves as a direct CAR ligand (Moore et al., 2000; Tzameli et al., 2000).
Xiong et al. (2002) conclude that CAR does not play a significant role in regulating the inducible expression of Mrp3 gene expression in rats. Other research indicates that in rats, the PXR activators PCN, spironolactone, and dexamethasone do not induceMrp3 gene expression (Cherrington et al., 2002). Our data indicate that in mice, selective activation of PXR or CAR regulates the inducible expression of Oatp2 and Mrp3. Our studies agree with another recent report that indicates in mice, PXR and CAR play critical roles in regulating Mrp3 gene expression (Maglich et al., 2002). Taken together, these data are consistent with the notion that nonliganded PXR plays a negative or competitive pharmacological role in regulating both drug metabolism and drug transport, possibly through active repression of or competition for enhancer elements within the promoters of shared PXR/CAR-target genes. The study we have presented here highlights the utility of PXR knockout mice in determining the role of PXR in regulating gene expression and activity in vivo.
Acknowledgments
This research was supported by the Centers of Biomedical Research Excellence grant in Protein Structure and Function National Institutes of Health Grant RR17708-01. We also acknowledge Dan Brobst for technical assistance.
Footnotes
- Abbreviations used are::
- P450s
- cytochromes P450
- PXR
- pregnane X receptor
- CAR
- constitutive androstane receptor
- PXR-KO
- PXR knockout mouse
- PCN
- pregnenalone 16α-carbonitrile
- PB
- phenobarbital
- Oatp2
- organic anion transporting polypeptide
- Mrp3
- multi-drug resistance associated protein 3
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- rtQ-PCR
- real-time quantitative polymerase chain reaction
- Received October 31, 2002.
- Accepted February 10, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}