


Abstract
O-Glucuronidation of 5-hydroxyrofecoxib is the major biotransformation pathway of rofecoxib in human, rat, and dog. The glucuronide conjugate is also involved in the reversible metabolism of rofecoxib in rat and human. Atypical bimodal phenomena were observed in their plasma concentration-time curves with a large variability among different human subjects. It is unclear which family members of human UDP-glucuronosyltransferases (UGT) are involved in the formation of the glucuronide. O-Glucuronidation of 5-hydroxyrofecoxib by human liver microsomes and eight cDNA-expressed human UGT isoforms were investigated. Human liver microsomes formed 5-hydroxyrofecoxib glucuronide with apparent Vmax value of 1736 pmol/min/mg of protein and Km value of 44.2 μM. Eight individual cDNA-expressed human UGT isozymes (1A1, 1A3, 1A4, 1A6, 1A8, 1A9, 2B7, and 2B15) were evaluated for glucuronidation of 5-hydroxyrofecoxib. Among them UGT2B15 exhibited the highest metabolism rate with apparent Vmax value of 286 pmol/min/mg of protein and Km value of 16.1 μM, whereas UGT2B7 showed apparentVmax value of 47.1 pmol/min/mg of protein and Km value of 41.6 μM. These results indicated that human UGT2B15 has the highest level of activity for catalyzing the glucuronidation of 5-hydroxyrofecoxib. Because polymorphisms have been identified in human UGT2B7, 2B15 genes andO-glucuronidation of 5-hydroxyrofecoxib plays a major role in biotransformation of rofecoxib, it is possible that human UGT2B7 and 2B15 polymorphisms for O-glucuronidation of 5-hydroxyrofecoxib are responsible for the high variability in bimodal patterns in human plasma concentration-time curves.
Rofecoxib, 3-phenyl-4-[4-(methylsulfonyl)phenyl]-2(5H)-furanone, is highly selective for inhibition of the inducible form of cyclooxygenase (COX-21) and is marketed by Merck as an anti-inflammatory drug (VIOXX) for the treatment of arthritis and pain (Chan et al., 1999; Prasit et al., 1999). The absorption, distribution, metabolism, and excretion of rofecoxib in rat, dog, and human have been reported (Halpin et al., 2000, 2002). Rofecoxib was extensively metabolized with the major metabolites as 5-hydroxyrofecoxib and its O-glucuronide conjugate. In the rat, an unusual feature of the plasma concentration versus time profile for rofecoxib following oral administration was the presence of a distinct second Cmax. Similar phenomena were also observed in human pharmacokinetic profiles with a large variability among different subjects. It was suggested that the atypical bimodal phenomena in plasma concentration-time curves were due to reversible metabolism of 5-hydroxyrofecoxib to rofecoxib (Baillie et al., 2001). The 5-hydroxyrofecoxib was metabolized to its glucuronide conjugate and excreted in bile. The glucuronide metabolite was deconjugated in the lower gastrointestinal tract, resulting in 5-hydroxyrofecoxib. Reduction of the 5-hydroxyrofecoxib formed a hydroxyacid that cyclized spontaneously to regenerate rofecoxib, which was reabsorbed and entered the systemic circulation. The secondCmax was due to reabsorption of rofecoxib formed from 5-hydroxyrofecoxib glucuronide in the lower gastrointestinal tract.
Since O-glucuronidation of 5-hydroxyrofecoxib plays a major role in biotransformation and reabsorbtion of rofecoxib in human, it is interesting to identify human UGT enzymes responsible for the glucuronidation of 5-hydroxyrofecoxib. To date no human UGT isoform responsible for the glucuronidation of 5-hydroxyrofecoxib has been reported. The primary goal of the present investigation was to determine which human UGT isoforms are responsible for the glucuronidation of 5-hydroxyrofecoxib and to understand the mechanism of high variability in pharmacokinetic patterns in human subjects. 5-Hydroxyrofecoxib was incubated with human liver microsomes and eight cDNA expressed human UGT isoforms from 1A and 2B subfamilies that are involved in xenobiotic metabolism (Mackenzie et al., 1997; Jedlitschky et al., 1999). The incubated samples were analyzed by LC-MS coupled with UV detection. For active UGT isoforms, kinetic parameters were determined and compared with those determined for human liver microsomes in an attempt to better explain the variation in clearance of the compound.
Materials and Methods
Chemicals.
[14C]Rofecoxib and 5-hydroxyrofecoxib were synthesized as described previously (Halpin et al., 2000; Baillie et al., 2001). Uridine diphosphoglucuronic acid (UDPGA), saccharic acid-1,4-lactone, alamethicin and β-glucuronidase (Helix Pomatia) were purchased from Sigma-Aldrich (St. Louis, MO). Pooled human liver microsomes were obtained from Xenotech (Kansas City, KS). Recombinant human UGT1A1, 1A3, 1A4, 1A6, 1A8, 1A9, 2B7, and 2B15 expressed in baculovirus-infected insect cells (Supersomes) were purchased from BD Gentest (Woburn, MA). 5-Hydroxyvaldecoxib was obtained from Pharmacia compound files. (Skokie, IL). All other chemicals and reagents were of analytical grade and available commercially.
Assay for Human Liver Microsomal UGTs.
In a typical incubation, 0.25 mg of human liver microsomes, 0.1 M potassium phosphate (pH 7.1), 5 mM saccharolactone, 50 mg alamethicin, 1 mM MgCl2, and 20 μM 5-hydroxyrofecoxib in a final volume of 0.5 ml were preincubated at 37°C for 10 min. To initiate the reaction, UDPGA (5 mM in incubation) was added and incubated at 37°C for 0 to 60 min in a shaking water bath. Control incubations were performed without human liver microsomes or without UDPGA. The reaction was stopped with 1 ml of 0.1% formic acid in ice-cold acetonitrile or methanol. The samples were centrifuged to remove precipitated protein and the supernatants were transferred and evaporated to dryness under nitrogen gas at room temperature. The samples were reconstituted in mobile phase A (see HPLC-UV andLC-MS) and analyzed by HPLC and LC-MS.
Assays for cDNA-Expressed Human UGT Isoenzymes.
All reaction mixtures contained 10 mM potassium-phosphate buffer (pH 7.1), 1 mM MgCl2, 5 mM saccharolactone, and 20 μM 5-hydroxyrofecoxib in a total volume of 0.5 ml. Expressed human UGT1A1, 1A3, 1A4, 1A6, 1A8, 1A9, 2B7, and 2B15 with a protein concentration of 0.5 mg/ml were added and preincubated at 37°C for 10 min. The reaction was initiated by the addition of 5 mM UDPGA and incubated at 37°C for 0 to 60 min in a shaking water bath. Control incubations were performed without UDPGA. The reaction was terminated with 1 ml of 0.1% formic acid in ice-cold acetonitrile or methanol. The samples were centrifuged to pellet precipitated protein. The supernatants were transferred and evaporated to dryness under nitrogen gas under room temperature. The samples were resolved in mobile phase A and analyzed by HPLC and LC-MS.
β-Glucuronidase Hydrolysis.
The O-glucuronide of 5-hydroxyrofecoxib produced from human liver microsomes as mentioned above was isolated by HPLC, dried under nitrogen gas, and reconstituted in 1.5 ml of 0.2 M sodium acetate buffer (pH 5.0). Aliquots (0.5 ml) of each sample were incubated in the absence (control) and presence of 200 units of β-glucuronidase (Sigma-Aldrich) for 16 h at 37°C in a shaking water bath. The incubations were stopped by the addition of 1 ml of acidified ice-cold methanol (0.1%). After evaporation to dryness under nitrogen gas, the samples were resolved in mobile phase A and analyzed by LC-MS.
Stability of O-Glucuronide of 5-Hydroxyrofecoxib.
5-Hydroxyrofecoxib glucuronide from in vitro incubations with human liver microsomes was resolved in 0.2 ml of each buffer, with specific pH values of 3, 6, 7, and 9. The resulting mixtures then were injected immediately onto the LC-MS system and re-injected at an interval of 2 h over a 6-h period after standing on an autosampler at room temperature. For each pH mixture, the time for the first injection was considered as the starting time (time = 0).
HPLC-UV and LC-MS.
HPLC-UV and LC-MS analyses were performed using an Agilent HPLC system (1100 series) linked to a Thermo Finnigan LCQ-Deca ion-trap mass spectrometer (San Jose, CA). The MS was equipped with an electrospray interface. The LC system consisted of an autosampler, an HPLC pump, and a diode-array detector (Agilent Technologies, San Fernando, CA). The separation was carried out on a Zorbax phenyl column (2.1 × 150 mm, 5 μm; Agilent Technologies) at ambient temperature with a linear gradient system that was programmed from 100% mobile phase A to 100% mobile phase B in 20 min, followed by isocratic condition of 100% mobile phase B for 5 min. The system then was programmed back to 100% mobile phase A in 1 min and re-equilibrated for 5 min before the next injection. The flow rate was 0.2 ml/min. Mobile phase A consisted of acetonitrile/water/formic acid (5:95:0.1, v/v/v), and mobile phase B consisted of acetonitrile/water/formic acid (20:80:0.1, v/v/v). The effluent from HPLC column passed through the diode-array detector, monitored at 280 nm, then introduced into the mass spectrometer. The mass spectral analysis was performed using electrospray ionization in negative ion mode. The source voltage and current were set up at 5 kV and 80 μA, respectively. The capillary voltage was set at −4 V, with the heated capillary temperature held at 200°C. The optimum collision energy for MS/MS was in the range of 20 to 30% of normalize collision energy. Three events were carried out for the analyses and these included MS, MS/MS of m/z 329 and MS/MS ofm/z 505. A divert time of 2 min was set at the start of each chromatographic run to prevent early eluting matrix material from blocking the heated capillary region of the MS system and contaminating the ion optic region.
Enzyme Kinetic Measurements.
The apparent enzyme kinetic parameters ofKm andVmax were determined for the glucuronidation of 5-hydroxyrofecoxib using human liver microsomes and expressed human UGT2B7 and 2B15 by varying the substrate concentration (0.75–100 μM) at a fixed concentration of UDPGA (5 mM). After incubations, internal standard 5-hydroxyvaldecoxib (50 μM) was added into the samples. The samples were extracted as described previously and the concentrations of 5-hydroxyrofecoxib glucuronide in the samples were quantitated by LC-MS/MS against its standard curve. Since the synthetic standard of 5-hydroxyrofecoxib glucuronide was not available, [14C]5-hydroxyrofecoxib glucuronide was isolated by HPLC from cyno monkey urine after oral administration of [14C]rofecoxib with specific activity of 3.16 μCi/mg (C. S. Cook and J. Y. Zhang, unpublished work) and used as the standard. The concentration of [14C]5-hydroxyrofecoxib glucuronide was quantitated based on the radioactive measurement [concentration = radioactivity × specific activity of the dose compoundX (molecular weight of 5-hydroxyrofecoxib glucuronide)/(molecular weight of the dose compound)]. The peak areas of 5-hydroxyrofecoxib glucuronide (m/z 505 →m/z 329) and internal standard (m/z 329 → m/z 196) generated by the LC-MS/MS were obtained from the Xcalibur data system (Thermo Finnigan). The ratios of the peak areas ofm/z 505 → m/z 329 tom/z 329 → m/z 196 were then calculated for 5-hydroxyrofecoxib glucuronide. The standard curve was obtained by a weighted (1/concentration2) least-squares linear regression analysis. Concentrations of 5-hydroxyrofecoxib glucuronide in the samples were calculated using the equations from the standard curve. Estimated apparent Km andVmax were obtained by fitting the glucuronidation data to an equation describing Michaelis-Menten kinetics by nonlinear regression analysis using WinNonlin standard for PC (Pharsight Corp., Mountain View, CA).
Results
Glucuronidation of 5-Hydroxyrofecoxib in Human Liver Microsomes.
5-Hydroxyrofecoxib was incubated with human liver microsomes in the absence and presence of UDPGA at 37°C up to 60 min. After the incubations, there was no observable turnover of 5-hydroxyrofecoxib in the absence of UDPGA. However, in presence of UDPGA a polar peak was observed in 16.8 min, 1.5-min earlier than 5-hydroxyrofecoxib (Fig.1). The major peak at 18.3 min had the same HPLC retention time as the 5-hydroxyrofecoxib standard. The LC-MS analysis showed that the peak had a deprotonated molecular ion [M − H]− at m/z 329, consistent with that of 5-hydroxyrofecoxib (Fig.2). The product ion spectrum ofm/z 329 generated major fragment ions atm/z 303, 285, and 257, which were formed by the loss of 26 (CH=CH), 44 (CO2), and 72 Da (COCO2) from [M − H]−, which were the same as that of 5-hydroxyrofecoxib standard (Fig. 3A). These results confirmed the major peak as the unchanged 5-hydroxyrofecoxib. The LC-MS analysis indicated that the polar peak at the retention time of 16.8 min had an [M − H]− ion at m/z 505, 176 mass units higher than that of 5-hydroxyrofecoxib, suggesting that it was a glucuronide conjugate of 5-hydroxyrofecoxib (Fig. 2). The product ion spectrum of m/z 505 generated fragment ions at m/z 329, 285, and 257 (Fig. 3B), which were formed by the loss of 176 (dehydroglucuronic acid), 44 (CO2), and 72 Da (COCO2) from m/z 505, suggesting that the site of glucuronide conjugation occurred at the 5-hydroxyl group of 5-hydroxyrofecoxib. Based on the MS data, the polar peak was identified as an O-glucuronide conjugate of 5-hydroxyrofecoxib. No glucuronide conjugate was observed in control incubations that lacked human liver microsomes or UDPGA, or the zero minute control. The results suggested that the 5-hydroxyrofecoxib glucuronide was formed enzymatically and was UDPGA-dependent.

HPLC-UV chromatograms derived from the incubations of 5-hydroxyrofecoxib (5-OH) with human liver microsomes in the absence (A) and presence (B) of UDPGA at 37°C for 60 min.
5-Hydroxyrofecoxib glucuronide (5-OH-G) was formed in the samples from incubation in presence of UDPGA.

LC-MS chromatograms derived from the incubations of 5-hydroxyrofecoxib with human liver microsomes in the absence (A) and presence (B) of UDPGA at 37°C for 60 min.

MS/MS spectra of 5-hydroxyrofecoxib (A) and 5-hydroxyrofecoxib glucuronide (B) in the samples from the incubations with human liver microsomes in presence of UDPGA.
Glucuronidation of 5-Hydroxyrofecoxib in cDNA-Expressed Human UGT Isoforms.
Eight commercially available human recombinant UGT isoforms from 1A and 2B families were examined for the formation of 5-hydroxyrofecoxib glucuronide conjugate. The results indicated that expressed human UGT1A9, 2B7, and 2B15 catalyzed the formation ofO-glucuronide conjugate of 5-hydroxyrofecoxib. In contrast, very little conjugate was detected in incubates with expressed UGT1A1, 1A3, 1A4, 1A6, and 1A8 (Table 1). Per milligram of protein, the rate of glucuronidation of 5-hydroxyrofecoxib by UGT2B15 (161.4 ± 35.9 pmol/min/mg, n = 3) was much higher than those of 2B7 (15.8 ± 3.48 pmol/min/mg,n = 3) and 1A9 (3.2 ± 1.3 pmol/min/mg,n = 3) under the experimental conditions used. These results clearly suggested that UGT2B15 is the most important isoform catalyzing the glucuronidation of 5-hydroxyrofecoxib.
Glucuronidation activity of 5-hydroxyrofecoxib by different UGT sources at 5 mM UDPGA
Kinetic Parameters of 5-Hydroxyrofecoxib Glucuronide in Human Liver Microsomes, UGT2B7 and 2B15.
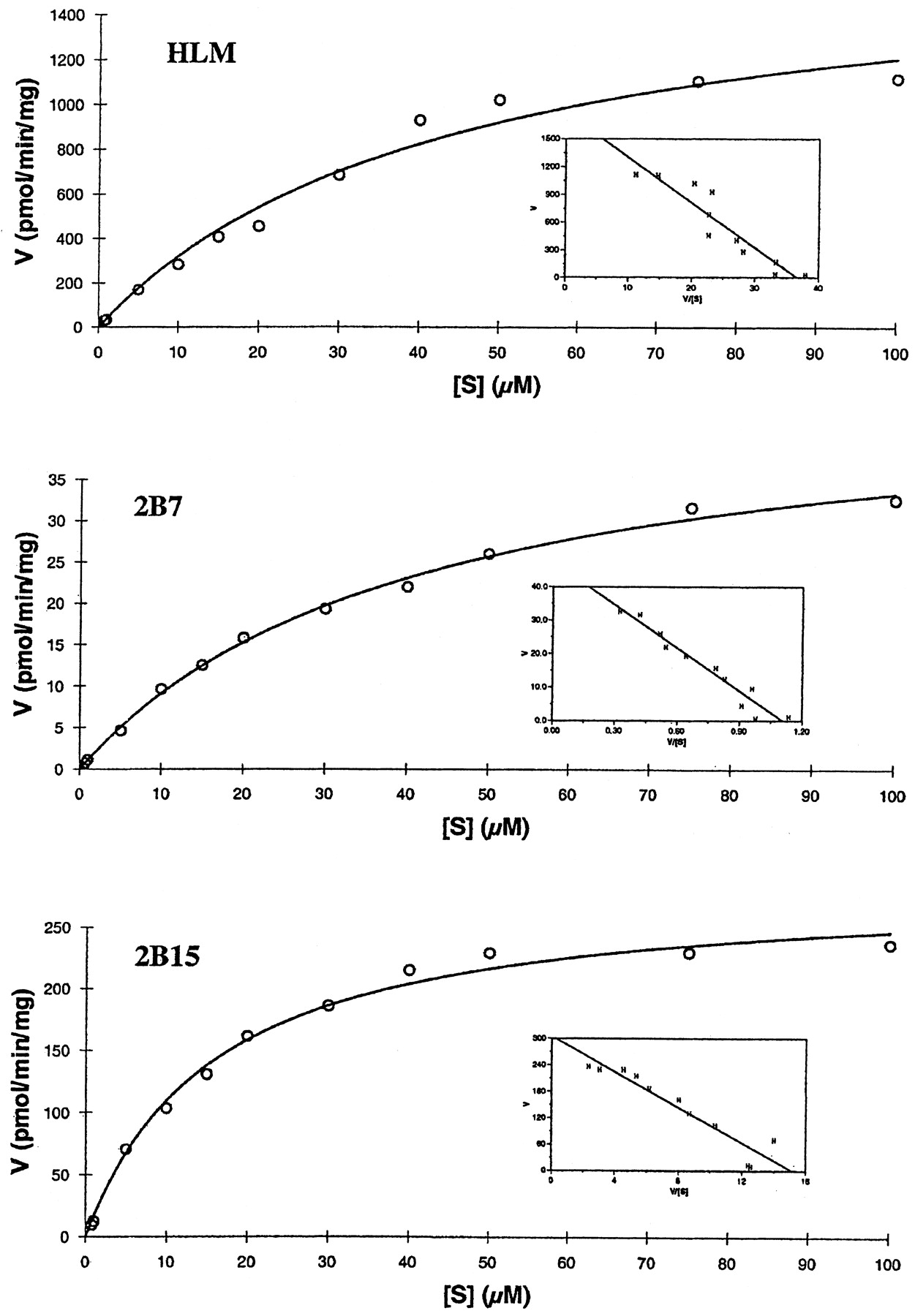
The formation rate of 5-hydroxyrofecoxib glucuronide as a function of substrate concentration was measured in pooled human liver microsomes and expressed UGT2B7 and 2B15 (Fig. 4). All three reactions followed single Michaelis-Menten kinetics. Nonlinear transformation of the data yielded a mean apparentKm andVmax of 44.2 ± 2.7 μM and 1736 ± 86.8 pmol/min/mg (n = 3) for human liver microsomes, respectively (Table 2). For expressed human UGT2B7 and 2B15, apparentKm andVmax values were 41.6 ± 10.4, 16.1 ± 3.2 μM and 47.1 ± 11.3, 286 ± 62.9 pmol/min/mg (n = 3), respectively. ApparentKm andVmax values for UGT1A9 were not measured due to its low glucuronidation activity. Catalytic efficiencies (Vmax/Kmratios) for human liver microsomes, expressed UGT2B7 and 2B15 were 39.3, 1.13, and 17.8 μl/min/mg, respectively. The apparent kinetic parameter (Vmax/Km) of 2B15 was much greater that that of UGT2B7, further demonstrating that UGT2B15 is a predominant isoform for the glucuronidation of 5-hydroxyrofecoxib in human liver. However, theVmax/Kmratio of UGT2B15 was approximately 50% of that of human liver microsomes. It is possible that this apparent difference may be due to some other UGT isoforms, which we have not tested, involved in the glucuronidation of 5-hydroxyrofecoxib in human liver microsomes.

Effect of substrate concentration on the rate of glucuronidation of 5-hydroxyrofecoxib by human liver microsomes (HLM), UGT2B7 and 2B15.
Apparent kinetic parameters for glucuronidation of 5-hydroxyrofecoxib by human liver microsomes, UGT2B7 and 2B15
Stability of O-Glucuronide Conjugate of 5-Hydroxyrofecoxib.
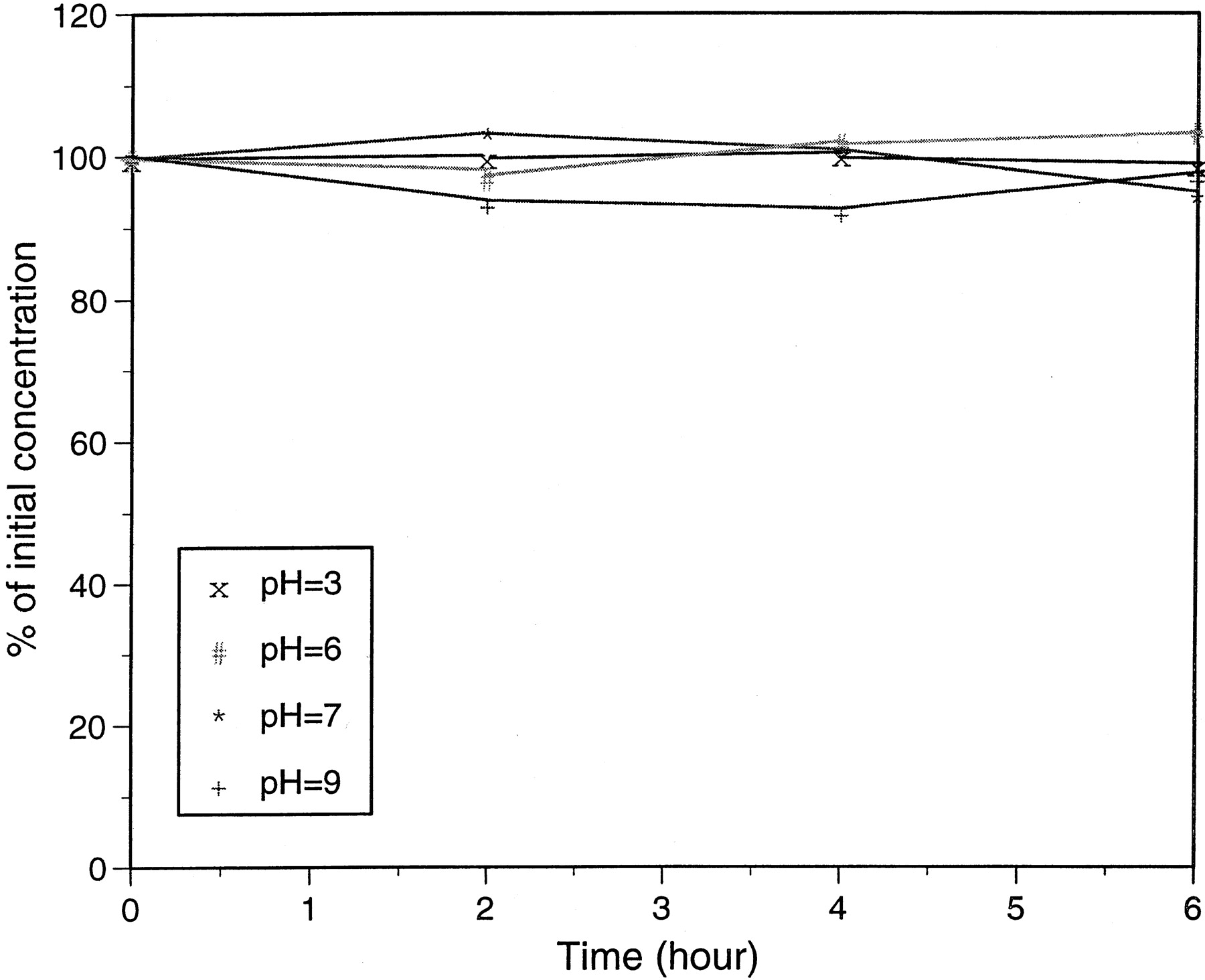
5-Hydroxyrofecoxib glucuronide isolated from in vitro incubation was stable under both acidic and basic conditions (pH 3 to 9) (Fig.5). No significant degradation of the conjugate was observed up to 6 h at room temperature under these pH conditions. The results suggested hydrolysis of the 5-hydroxrofecoxib glucuronide is unlikely catalyzed chemically.

Stability of 5-hydroxyrofecoxib glucuronide conjugate as a function of time in different pH buffers at room temperature.
Hydrolysis of 5-Hydroxyrofecoxib Glucuronide by β-Glucuronidase.
When 5-hydroxyrofecoxib glucuronide was incubated with β-glucuronidase, it was hydrolyzed and converted to 5-hydroxyrofecoxib. The hydrolysis was effectively inhibited by the specific β-glucuronidase inhibitor d-saccharic acid-1,4-lactone, confirming that the hydrolysis of the conjugate is enzyme-dependent.
Discussion
Rofecoxib is metabolized in human to 5-hydroxyrofecoxib and itsO-glucuronide conjugate, but until now, no human UGT has been identified to catalyze the formation of the 5-hydroxyrofecoxib glucuronide. We have investigated the formation ofO-glucuronide of 5-hydroxyrofecoxib using human liver microsomes and cDNA-expressed human UGT isoforms. We confirmed the formation of O-glucuronide conjugate of 5-hydroxyrofecoxib after the incubation of 5-hydroxyrofecoxib with human liver microsomes and UGPGA by the LC-MS/MS analyses. We also demonstrated that three UGT isoforms 1A9, 2B7, and 2B15 among eight isoforms that we tested exhibited measurable catalytic activities in metabolism of 5-hydroxyrofecoxib. UGT2B15 exhibited the highest metabolism rate, almost 15-fold higher than UGT2B7, whereas UGT1A9 showed very low activity. The apparentVmax/Kmvalues of human liver microsomes, UGT2B7 and UGT2B15 were 39.3, 1.13 and 17.8 μl/min/mg, respectively. These results suggest that UGT2B7 and 2B15 isoforms are the major enzymes involved in theO-glucuronidation of 5-hydroxyrofecoxib in human liver microsomes.
The UGTs are a superfamily of membrane-bound enzymes that catalyze the conjugation of endo- and xenobiotics with d-glucuronic acid. Currently, over 30 human UGT genes have been identified and classified into two families (UGT1 and UGT2) based on their sequence similarities. Among these human UGT genes, only five UGT genes (UGT1A1, UGT1A6, UGT2B4, UGT2B7, and UGT2B15) have been described as polymorphisms (Mackenzie et al., 2000). UGT2B7 is a major UGT in the liver and gastrointestinal tract. It has the capability to glucuronidate a range of drugs including morphine and nonsteroidal anti-inflammatory agents (Ritter et al., 1990; Jin et al., 1993;Coffman et al., 1997). Two UGT2B7 variants with either His or Tyr at position 268 have been identified (Jin et al., 1993). UGT2B15 has activity toward several classes of flavonoids, drugs, and steroids (Green et al., 1994). Two polymorphic alleles that encode UGT2B15 variants with either Tyr or Asp at position 85 have been identified (Levesque et al., 1997). UGT2B15 is expressed in numerous human tissues such as liver, kidney, testis, mammary gland, prostate, and lung. Significant ethnic differences between Asians and Caucasians in the distribution of these polymorphisms in UGT2B7 and 2B15 have been observed (Lampe et al., 2000). It was also reported that homozygous, lower-activity Asp85 allele of UGT2B15 was significantly more common in prostate cancer patients than in control individuals (MacLeod et al., 2000). Furthermore, polymorphisms in the UGTs have been postulated to contribute to interindividual variation in drug disposition (Yue et al., 1989; Patel et al., 1995). However, there was little evidence that these polymorphisms have any major clinical significance unless the UGT in question is responsible for the exclusive metabolism of a particular drug or chemical (Mackenzie et al., 2000).
In clinical studies, the plasma pharmacokinetic profiles of rofecoxib exhibited very unusual bimodal phenomena with twoCmax values presented in its plasma concentration versus time profiles following oral administration. In addition, a substantial variability for the secondary peaks was observed in different human subjects with at least four types of shapes detected (C. S. Cook, unpublished work). The presence of these secondary peaks was reflected in the variability observed fortmax (Halpin et al., 2002). A similar phenomenon was also observed in the rats with no variability in the secondary peak. It has been suggested that the atypical bimodal phenomena in plasma concentration-time curves were due to reversible metabolism of 5-hydroxyrofecoxib to rofecoxib (Baillie et al., 2001). The reversible mechanism was proposed as shown in Fig.6. The 5-hydroxyrofecoxib was metabolized to its glucuronide conjugate and excreted in bile. The glucuronide metabolite was then deconjugated in the lower gastrointestinal tract, resulting in the formation of 5-hydroxyrofecoxib. The ring opening and reduction of the 5-hydroxyrofecoxib formed a hydroxyacid that cyclized spontaneously to regenerate rofecoxib, which was reabsorbed and entered the systemic circulation. The secondCmax was due to reabsorption of rofecoxib formed from 5-hydroxyrofecoxib glucuronide in the lower gastrointestinal tract. The reversible metabolism of 5-hydroxyrofecoxib to rofecoxib was confirmed by the measurement of rofecoxib after oral administration of 5-hydroxyrofecoxib in rat and human. In addition, when rofecoxib was administered to cholecystectomy patients, the corresponding profiles did not show the secondaryCmax and the absolute concentrations of rofecoxib in the cholecystectomy patients were approximately 20% of those observed in the plasma from healthy subjects (Halpin et al., 2002). These results clearly suggested the involvement of enterohepatic recycling via glucuronidase-catalyzed hydrolysis and subsequent intestinal reabsorption of the drug in human. Based on the above mechanism, glucuronidation of 5-hydroxyrofecoxib plays a critical role in the reversible metabolism of rofecoxib. Our studies have demonstrated that UGT2B7 and 2B15 were the major UGT isoforms involved in the glucuronidation of 5-hydroxyrofecoxib. Since genetic polymorphisms have been identified in human UGT2B7 and 2B15 genes, the polymorphisms of UGT2B7 and 2B15 for O-glucuronidation of 5-hydroxyrofecoxib may explain the phenomena of substantial variability in human plasma concentration versus time profiles and the difference between human and rats. From a mechanistic standpoint, the polymorphisms of UGT2B7 and 2B15 in different human subjects may have the different catalytic efficiency for the formation of 5-hydroxyrofecoxib glucuronide, which finally affects the concentrations of the reabsorbed 5-hydroxyrofecoxib and subsequent rofecoxib in the plasma, resulting in the variability in the secondaryCmax peak (Fig. 6). Furthermore, the lack of polymorphisms of UGTs in rats may explain the absence of the pharmacokinetic variability phenomenon in rats. To further address this issue, the correlation between the interindividual differences in rofecoxib pharmacokinetic profiles and genotypes of the UGT2B7 and 2B15 in these human subjects needs to be investigated. If the correlations were valid, the genetic polymorphisms of UGT2B7 and 2B15 genes for metabolism of rofecoxib would be clinically important. Therefore, the pharmacokinetic profile variability between different subjects, especially different ethnic groups, due to the genetic polymorphisms of UGT2B7 and 2B125 may need to be considered for prescribing the drug. Moreover, possible drug-endobiotic interaction must be considered when rofecoxib is administered with such substances that also interact with UGT2B7 and 2B15, such as catechol estrogens and dihydrotestosterone. Drug-drug interactions should also be considered, because UGT2B7 and 2B15 are major isoforms catalyzing glucuronidations of nonsteroidal anti-inflammatory drugs, benzodiazepine, morphine, epirubicin, and coumarins, etc. Further studies on the substrate specificity and kinetic analyses for genetic polymorphisms of UGT2B7 and 2B15 need to be investigated to identify possible drug-endobiotic and/or drug-drug interactions.

Proposed mechanism for the enterohepatic cycling of rofecoxib via 5-hydroxyrofecoxib and its glucuronide
In summary, our study has identified UGT2B7 and 2B15 as the major isoforms involved in the glucuronidation of 5-hydroxyrofecoxib in human liver microsomes. Because polymorphisms have been identified in human UGT2B7 and 2B15 genes and O-glucuronidation of 5-hydroxyrofecoxib plays a major role in reversible metabolism of rofecoxib in humans, it is possible that human UGT2B7 and 2B15 polymorphisms for O-glucuronidation of 5-hydroxyrofecoxib are responsible for the high variability in bimodal patterns in human plasma concentration-time curves. However, the clinical significance of UGT2B7 and 2B15 polymorphisms in the metabolism of rofecoxib needs to be further investigated.
Acknowledgments
We thank Jennifer Willard for skillful technical assistance and help with preparation of the manuscript.
Footnotes
- Abbreviations used are::
- COX-2
- cyclooxygenase II
- UGT
- UDP-glucuronosyltransferases
- UDPGA
- UDP-glucuronic acid
- HPLC
- high performance liquid chromatography
- LC-MS
- liquid chromatography-mass spectrometry
- Received December 23, 2002.
- Accepted February 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}