


Abstract
Troglitazone is a thiazolidinedione insulin sensitizer drug that is metabolized mainly to a sulfate conjugate (M-1) in humans. It was reported to cause hepatotoxicity, although the cause has not been fully clarified. The objective of this study was to identify whether organic anion transporting polypeptide (OATP) transporters expressed at the basolateral membrane of human hepatocytes participate in troglitazone-associated hepatotoxicity. When OATP-B, OATP-C, or OATP8 was expressed in Xenopus oocytes, the transporter-mediated uptake into oocytes of troglitazone sulfate conjugate and the inhibitory effects of thiazolidinediones and the metabolites of troglitazone on estrone-3-sulfate transport were measured. M-1 was transported well by OATP-C but was not transported by OATP-B. OATP8 showed weak, but not statistically significant, transport of M-1. M-1 exhibited a strong inhibitory effect on estrone-3-sulfate transport by OATP-C and OATP8, suggesting a higher affinity than other thiazolidinediones and the metabolites of troglitazone, glucuronide conjugate and quinone metabolite. In conclusion, the sulfate conjugate of troglitazone has a higher affinity for OATPs than troglitazone itself or other metabolites. Since OATP transporters are important in the hepatic handling of bile acids, bilirubin, and other endogenous anionic compounds, M-1 may disturb the hepatic influx and efflux transport of these endogenous molecules across the basolateral membranes. Moreover, OATP-C may be involved in the hepatic toxicity of troglitazone through the inhibitory action of M-1.
Troglitazone (Fig. 1) is a thiazolidinedione insulin sensitizer drug for the treatment of type 2 noninsulin-dependent diabetes mellitus. Despite its effectiveness, it was withdrawn from the market because of hepatic toxicity. Although there have been several studies investigating the mechanisms of the hepatotoxicity, including apoptotic hepatocyte death and intrahepatic cholestasis, the details remain to be clarified (Gitlin et al., 1998; Kostrubsky et al., 2000; Yamamoto et al., 2001). In humans, the metabolism of troglitazone involves sulfation, glucuronidation, and oxidation (Kawai et al., 1997; Loi et al., 1999). The sulfate conjugate (M-1; Fig. 1) is the most abundant metabolite, and the glucuronide conjugate (M-2; Fig. 1) and quinone metabolite (M-3; Fig. 1) are relatively minor (Loi et al., 1999). After i.v. administration of troglitazone to rats, the concentration of M-1 in plasma and liver reached about 10 times that of troglitazone itself (Funk et al., 2001). Therefore, M-1 might be involved in troglitazone-associated hepatotoxicity.

Chemical structures of troglitazone, its metabolites, pioglitazone, and rosiglitazone.
After oral administration, cumulative recovery of troglitazone and its metabolites was 85% in feces (Loi et al., 1999), and M-1 excreted into bile was reabsorbed in the intestine as M-1 and troglitazone after deconjugation (Kawai et al., 2000). Moreover, the plasma area under the curve value of M-1 was 4-fold high in patients with impaired liver function than in healthy volunteers, and plasma half-life was longer in patients with moderate or severe hepatic impairment (Ott et al., 1998). Accordingly, the failure of hepatic excretion of M-1 might lead to hepatotoxicity, although M-1 itself is pharmacologically inactive and did not exhibit cytotoxicity in human hepatoma cells (Loi et al., 1999; Yamamoto et al., 2001).
Recently, it was reported that M-1 inhibited the hepatobiliary elimination of bile salts by the bile salt export pump (Bsep1) located on the canalicular membrane of hepatocytes in rats (Funk et al., 2001). Moreover, Kostrubsky et al. (2001) suggested that Mrp2, which is expressed on the canalicular membrane of hepatocytes, might participate in the excretion of troglitazone and its metabolites. When M-1 is accumulated in hepatocytes at high concentrations, it may disturb the hepatobiliary export of bile acids by inhibition of Bsep and Mrp2, leading to intrahepatic cholestasis. Therefore, in this study we focused on the hepatic basolateral membrane transporters, since they are important in the hepatic handling of physiological and xenobiotic compounds between blood and hepatocytes.
In the human, organic anion transporting polypeptides OATP-B (SLC02B1), OATP-C (SLC01B1), and OATP8 (SLC01B3) are expressed on the basolateral membrane of hepatocytes (Hagenbuch and Meier, 2003). Among them, OATP-C and OATP8 show a broad recognition spectrum for amphipathic substrates and play physiologically important roles in hepatic handling of xenobiotics and endogenous compounds.
We hypothesized that the troglitazone-associated hepatotoxicity is related to the accumulation of M-1 in hepatocytes by OATP transporters. Therefore, we examined the transport activity of OATP transporters for M-1 and the potential interaction of thiazolidinediones and/or the metabolites of troglitazone and endogenous OATP substrates.
Materials and Methods
Materials. [3H]Estrone-3-sulfate, ammonium salt (1961 GBq/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). Thiazolidinediones and the metabolites of troglitazone were kindly provided by Sankyo Co. (Tokyo, Japan).
All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and Wako Pure Chemicals (Osaka, Japan). cDNA Cloning of OATP Transporters. Cloning of cDNAs for OATP-B and OATP-C was described previously (Tamai et al., 2000). For cDNA cloning of OATP8, amplification of the coding sequence was performed by using human liver Marathon-Ready cDNA (BD Biosciences Clontech, Palo Alto, CA). Oligonucleotide primers (5′ primer: CGTGGATCCTGTAGTTTAATAATGGACC; 3′ primer: GTTGGAATTGTCAGTGAAAGACC) were designed based on the human OATP8 sequence (GenBank accession no. AJ251506). The polymerase chain reaction was performed with KOD (Plus) (Toyobo Engineering, Osaka, Japan) as follows: 2 min at 94°C, followed by 35 cycles of 15 s at 94°C,30 s at 60°C, and 2.5 min at 68°C. A major 2.2-kilobase polymerase chain reaction product was fully sequenced and subcloned into expression vector pcDNA3 (Invitrogen, San Diego, CA).
Transport Study. For transport experiments, oocytes were injected with complementary RNA (cRNA) of each OATP as described previously (Tamai et al., 2001). Uptake was initiated by incubating the oocytes at 25°C in modified Barth's solution (MBS; 96 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, and 10 mM HEPES, adjusted to pH 7.4 with NaOH) containing M-1 or [3H]estrone-3-sulfate. For measurement of M-1 uptake, 10 oocytes were washed and sonicated in 100 μl of MBS at appropriate times. The sample was treated with an equal volume of methanol and the precipitated protein was removed by centrifugation (13,000g, 10 min). A 100-μl portion of the supernatant was subjected to high-pressure liquid chromatography as described previously (Watanabe et al., 2002). For the uptake of [3H]estrone-3-sulfate, oocytes were washed and solubilized. Then, the associated radioactivity was measured by means of a liquid scintillation counter. The thiazolidinedione chemicals dissolved in dimethyl sulfoxide (10 mM stock solution) were used, and final concentration of the dimethyl sulfoxide was 0.1%.
Data Analysis. The uptake of test compound by OATP was evaluated after subtracting the uptake by uninjected oocytes from the uptake by OATP cRNA-injected oocytes. All data were expressed as means ± S.E.M., and statistical analysis was performed by Student's t test or Kruskal-Wallis test. The criterion of significance was p < 0.05.
Results
Uptake of M-1 by Oocytes Expressing OATP-B, OATP-C, or OATP8. To determine the transport activity of OATP-B, OATP-C, and OATP8 for M-1, the uptake of M-1 was examined at 10 μM using oocytes injected with cRNA of each OATP and compared with that by uninjected oocytes. The results are shown in Fig. 2. Since the uptake of M-1 by uninjected oocytes was equal to that by oocytes injected with 50 nl of water (data not shown), uninjected oocytes were used as the reference instead of water-injected oocytes in this study. OATP-C-expressing oocytes showed significantly increased uptake of M-1 compared with that by uninjected oocytes, whereas the uptake by OATP-B-expressed oocytes was comparable with that by uninjected oocytes. The uptake of M-1 by OATP8-expressed oocytes showed a tendency to increase compared with that by uninjected oocytes, but the difference was not statistically significant.

Uptake of troglitazone sulfate by oocytes injected with cRNA of OATP-B, OATP-C, or OATP8.
Oocytes injected with cRNA of OATP-B, OATP-C, or OATP8 (closed column) or uninjected (open column) were incubated at 25°C for 120 min in MBS (pH 7.4) containing troglitazone sulfate (10 μM). Uptake was expressed as cell-to-medium ratio. Each column represents the mean ± S.E.M. (n = 6–14), and * indicates a significant difference from the uptake by uninjected oocytes (p < 0.05).
Affinity of Troglitazone, its Metabolites, Pioglitazone, and Rosiglitazone, for OATP-C or OATP8. We evaluated the inhibitory effects of thiazolidinediones and the metabolites of troglitazone on the uptake of [3H]estrone-3-sulfate by OATP-C and OATP8. Since the plasma concentration of troglitazone has been reported to be 3.6 to 6.3 μM (Loi et al., 1997), we conducted the concentration of inhibitor at 1 and 10 μM as clinically relevant concentrations. Uptake of [3H]estrone-3-sulfate was significantly higher in oocytes injected with cRNA of both OATP-C and OATP8 than that by uninjected oocytes (Fig. 3). The uptake of [3H]estrone-3-sulfate by OATP-C reached 3.3 ± 0.3 μl/oocyte/30 min as a cell-to-medium ratio, whereas that by OATP8 was 0.79 ± 0.10 μl/oocyte/120 min, and that by noninjected oocytes was 0.17 ± 0.01 μl/oocyte/120 min. Figure 4 shows the inhibitory effect of troglitazone and its metabolites, pioglitazone and rosiglitazone, on the uptake of [3H]estrone-3-sulfate by OATP-C or OATP8. M-1 showed the strongest inhibitory effect on the uptake of [3H]estrone-3-sulfate by both OATP-C and OATP8, and its effect was concentration-dependent at 1 and 10 μM. M-2 inhibited only the uptake by OATP-C, but not that by OATP8. Pioglitazone and rosiglitazone significantly inhibited the uptake of [3H]estrone-3-sulfate by OATP-C, but their inhibitory effects were weaker than that of troglitazone. The uptake by OATP8 was significantly inhibited by pioglitazone but not by rosiglitazone at 10 μM. These results demonstrated that M-1 and related compounds have higher affinity for OATP-C than for OATP-8.
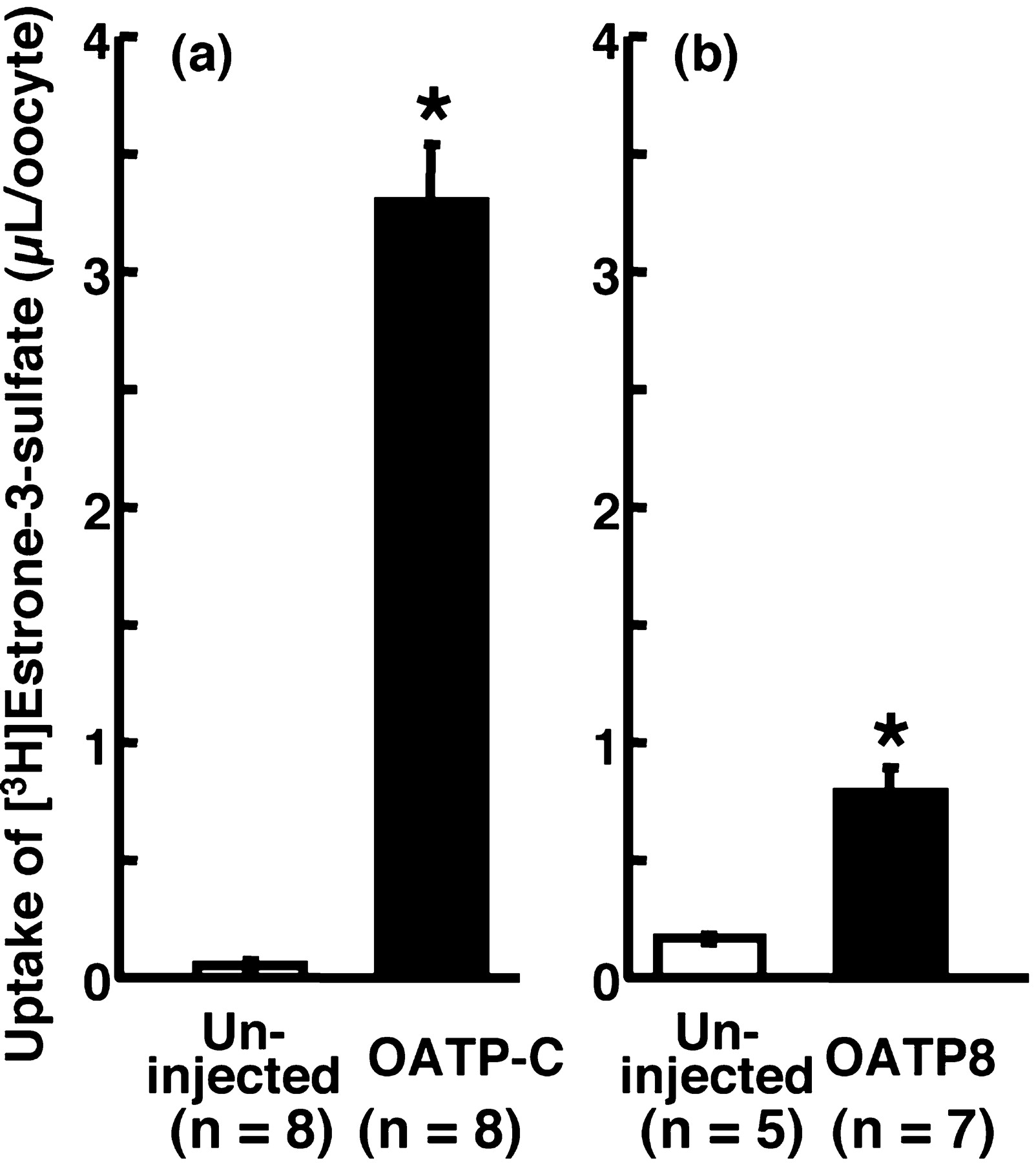
Uptakes of [3H]estrone-3-sulfate by oocytes injected with cRNA of OATP-C (a) or OATP8 (b).
Uptake of [3H]estrone-3-sulfate (3.6 nM) was measured at 25°C for 30 min (a) or 120 min (b). Open and closed columns represent the results in uninjected and OATP-C (a) or OATP8 (b) cRNA-injected oocytes, respectively. Each column represents the mean ± S.E.M. (n = 5–8), and * indicates a significant difference from uptake by uninjected oocytes (p < 0.05).

Inhibitory effects of several compounds on uptake of [3H]estrone-3-sulfate by oocytes expressing OATP-C or OATP8.
Oocytes injected with cRNA of OATP-C (a) or OATP8 (b). Uptake of [3H]estrone-3-sulfate (9.2 nM) was measured at 25°C for 30 min (a) or 120 min (b). The results are shown as a percentage of control uptake measured in the absence of inhibitor after subtracting uptake by uninjected oocytes (OATP-C, 3.3 ± 0.25 μl/oocyte/30 min and uninjected oocyte, 0.056 ± 0.013 μl/oocyte/30 min; OATP8, 0.22 ± 0.071 μl/oocyte/120 min and uninjected oocyte, 0.076 ± 0.0021 μl/oocyte/120 min). The inhibitor concentrations are indicated by the numbers in the figures. Each column represents the mean ± S.E.M. (n = 6–8), and * indicates a significant difference from the control (p < 0.05).
Discussion
Troglitazone is mainly metabolized to M-1, which is present at much higher concentration than troglitazone itself in plasma and liver after i.v. administration (Funk et al., 2001). Recently, Yamamoto et al. (2001) suggested that M-1 did not exhibit cytotoxicity in HepG2 cells and that M-1 plays a protective role in hepatocytes, whereas troglitazone itself and M-3 exhibited cytotoxic and apoptotic effects (Kostrubsky et al., 2000). However, although M-1 is pharmacologically and toxicologically inactive in cultured cells, the substantial accumulation of M-1 in plasma and hepatocytes may cause some interaction with endogenous and/or xenobiotic compounds at membrane transporters in vivo.
In the present study, we examined whether OATP transporters are involved in the accumulation of M-1 in human liver and whether thiazolidinediones and the metabolites of troglitazone inhibit OATP transporters expressed at the basolateral membrane of human hepatocytes. The uptake study, which used oocytes expressing OATP-B, OATP-C, or OATP8, demonstrated that OATP-C and possibly OATP8 could transport M-1, although we cannot conclude that OATP-B does not transport M-1 because of the less expressed activity of OATP-B in the oocytes in the present study (Fig. 2). As expected from previous studies that sulfate conjugates have higher affinity to OATP-C compared with glucuronide conjugates (Tamai et al., 2001), the uptake of [3H]estrone-3-sulfate by OATP-C was strongly inhibited by 1 and 10 μM M-1, to 14.5 and 5.2% of the control, respectively (Fig. 4a). Moreover, the inhibitory effect was stronger than that of sulfobromophthalein and cyclosporine, which inhibited the OATP-C-mediated [3H]estrone-3-sulfate uptake to 8.0 and 6.1% at 50 and 10 μM, respectively (Nozawa et al., 2003). Although the Km value of M-1 transport by OATP-C could not be determined in this study because of the limit of quantification by high-pressure liquid chromatography, the strong inhibition of M-1 suggested that M-1 has high affinity to OATP-C among the substrates of OATP-C (Hagenbuch and Meier, 2003). Therefore, M-1 would be taken up into human hepatocytes via OATP-C with high affinity and would be accumulated, leading to disturbance of the function of Bsep and Mrp2 (Funk et al., 2001; Kostrubsky et al., 2001).
Many test compounds inhibited [3H]estrone-3-sulfate uptake by OATP-C and OATP8 at 10 μM or at a lower concentration (Fig. 4). Since OATP-C and OATP8 have a broad spectrum of substrates, the inhibitory effects may result in some alteration of physiological homeostasis (Hagenbuch and Meier, 2003). In particular, it was reported that the plasma concentration of M-1 reached 2 to 10 μM after i.v. administration in rats and dogs at a dose of 5 mg/kg (Kawai et al., 1997). Accordingly, the M-1 concentration in plasma may be high enough to inhibit OATP transporters nearly completely.
Recently, the polymorphisms of transporters and enzymes have been reported to cause interindividual alterations of their expression levels and/or functions. Since troglitazone-associated hepatitis is thought to be rare and idiosyncratic, the hepatitis may be related to the genetic polymorphisms of OATP transporters. Some types of genetic polymorphism with functional alteration of OATP-C have been reported, and such alterations of apparent OATP-C activity may lead to accumulation of M-1 in plasma or liver, resulting in troglitazone-associated toxicity (Tirona et al., 2001; Michalski et al., 2002; Nozawa et al., 2002).
In conclusion, we demonstrated that OATP-C and possibly OATP8 transport the major metabolite of troglitazone, M-1, which could thereby be accumulated in human liver. Furthermore, thiazolidinediones and the metabolites of troglitazone inhibit OATP-C and/or OATP8, and the inhibitory effects may disturb the homeostasis of endogenous and/or xenobiotic compounds. These effects might account, at least in part, for the hepatic toxicity of troglitazone.
Footnotes
-
↵1 Abbreviations used are: Bsep, bile salt export pump; OATP, organic anion transporting polypeptide; cRNA, complementary RNA; MBS, modified Barth's solution.
-
This investigation was supported in part by The Mochida Memorial Foundation for Medical and Pharmaceutical Research and by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology.
- Received July 21, 2003.
- Accepted November 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}