


Abstract
Some inhibitory agents against CYP2B6 have been reported, but none of these has been extensively characterized or compared with others, as to the potency and selectivity of inhibition toward CYP2B6. The goal of this work was to find a selective and potent chemical in vitro inhibitor toward CYP2B6 using bupropion hydroxylation as a model reaction. At the initial screening of more than 30 substances, ticlopidine, triethylenethiophosphoramide (thioTEPA), metyrapone, xanthate C8, and benzylisothiocyanate displayed IC50 values of <10 μM and were selected for a more detailed analysis. Metyrapone, xanthate C8, and benzylisothiocyanate inhibited several other cytochrome P450 activities rather effectively, some of them even more potently than CYP2B6, and consequently are unsuitable as CYP2B6-selective probes. Ticlopidine and thioTEPA were the most potent inhibitors of bupropion hydroxylation with Ki values of 0.2 and 2.8 μM, respectively. The inhibition type of ticlopidine was found to be mixed type, with a component of mechanism-based inhibition, whereas thioTEPA inhibited CYP2B6 in a competitive manner. In addition to CYP2B6, ticlopidine also inhibited both mephenytoin 4-hydroxylation (CYP2C19) (IC50, 2.7 μM) and dextromethorphan O-demethylation (CYP2D6) (IC50, 4.4 μM). For thioTEPA the next sensitive P450 activity after CYP2B6 was coumarin 7-hydroxylation (IC50, 256 μM). Thus, although both compounds proved to be relatively potent inhibitors of CYP2B6, thioTEPA was about 2 orders of magnitude more selective than ticlopidine. Thus, thioTEPA is a drug of choice when high CYP2B6 selectivity among major P450 enzymes is required. Ticlopidine is a useful alternative under a controlled experimental setup and when higher potency is needed.
The identification of specific P4502 enzymes responsible for the metabolism of new chemical entities is important in the development of potential drugs. Together with the knowledge of the conditions that influence the expression and/or catalytic activity of the enzyme, predictions can be made as to whether or not environmental, medicinal, nutritional, chemical, or genetic factors could affect the metabolism of a new drug. Predictions can also be made on potential drug-drug interactions (induction or inhibition). The specific P450 enzyme(s) involved in the biotransformation of a compound can be a defining characteristic of its pharmacokinetic behavior.
The role of CYP2B6 in drug metabolism has not yet been thoroughly clarified. Although CYP2B6 represents only about 1 to 6% of the total P450 content in the human liver (Shimada et al., 1994; Stresser and Kupfer, 1999) there are a number of important substrates biotransformed mainly or partially by CYP2B6 in vitro (Mimura et al., 1993; Ekins et al., 1999; Gervot et al., 1999; Hanna et al., 2000; Hesse et al., 2000). Substrates and reactions claimed to be relatively specific to CYP2B6 include S-mephenytoin N-demethylation (Heyn et al., 1996; Ko et al., 1998; Kobayashi et al., 1999), 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide (RP 73401)-hydroxylation (Stevens et al., 1997; Domanski et al., 1999), 7-ethoxytrifluoromethylcoumarin O-de-ethylation (Code et al., 1997; Guo et al., 1997), methoxychlor ortho-hydroxylation (Stresser and Kupfer, 1999), bupropion hydroxylation (Faucette et al., 2000; Hesse et al., 2000), selegiline N-demethylation and N-depropargylation (Hidestrand et al., 2001), propofol oxidation (Oda et al., 2001), and efavirenz 8-hydroxylation (Ward et al., 2003). The studies of Faucette et al. (2000) and Hesse et al. (2000) demonstrated that bupropion hydroxylation is a selective model reaction for CYP2B6. Bupropion has been used as an antismoking agent, and understanding the metabolism of bupropion and in vitro interactions with other xenobiotics may give insight into the risk of adverse effects and potential interactions between bupropion and other drugs (Hesse et al., 2000).
Some inhibitory agents against CYP2B6 have been sufficiently characterized as to the potency and selectivity of inhibition toward CYP2B6. These include orphenadrine (Ekins et al., 1997; Guo et al., 1997), xanthates (Kent et al., 1999; Yanev et al., 1999), 2-phenyl-2-(1-piperidinyl)propane (Chun et al., 2000), ritonavir, efavirenz, and nelfinavir (Hesse et al., 2001). Recently, Piver et al. (2003) demonstrated that ϵ-viniferin is a potent mechanism-based inhibitor using 7-benzoxyresorufin-O-debenzoyloxylation as a model reaction for CYP2B6, and Rae et al. (2002) showed that thioTEPA (triethylenethiophosphoramide) is a selective inhibitor of CYP2B6 catalyzed S-mephenytoin N-demethylation to nirvanol. The work of Richter et al. (2004) was the first to report the effect of clopidogrel and ticlopidine on bupropion hydroxylation.
The goal of this work was to find a selective and potent chemical in vitro inhibitor of CYP2B6 using bupropion hydroxylation as the model reaction. More than 30 different chemicals were screened with respect to potency and extent of inhibition. The most potent CYP2B6 inhibitors were selected for more detailed investigations with human liver microsomes, recombinant expressed enzymes and P450-specific model reactions. Ticlopidine and thioTEPA were found to be the most selective and potent inhibitors of CYP2B6; thus, they can be used in in vitro P450 assignment studies and as reference inhibitors.
Materials and Methods
Chemicals. Bupropion [(±)-1-(3-chlorophenyl)-2-[(1,1-dimethylethyl) amino]-1-propanone] and hydroxybupropion (the exact site of the hydroxyl group has not been assigned) were donated by Glaxo SmithKline (Uxbridge, Middlesex, UK). Xanthate C8 was a generous gift from Prof. Stanislav Yanev, Department of Drug Toxicology, Medical University, Sofia, Bulgaria. Deramciclane, entacapone, nitecapone, selegiline, and tolcapone were obtained from Orion Pharma (Turku, Finland), ospemifene was purchased from Hormos Medical Corporation Ltd. (Turku, Finland), and troglitazone was obtained from Sankyo Pharmaceuticals (Tokyo, Japan). Midazolam and 1′-hydroxymidazolam were donated from F. Hoffman-La Roche (Basel, Switzerland), and rac-mephenytoin was donated from Novartis (Basel, Switzerland). Diethyldithiocarbamate, ethynylestradiol, ketoconazole, l-methamphetamine, methimazole, metyrapone, nicotine, orphenadrine, pyridine, quercetin, quinidine, retinol, sulfaphenazole, tamoxifen, thioTEPA, and tranylcypromine were purchased from Sigma-Aldrich (St. Louis, MO), ticlopidine was purchased from MP Biomedicals (Irvine, CA), and benzylisothiocyanate was purchased from Fluka (Buchs, Switzerland). Furafylline and the metabolite standards dextrorphan, 6-hydroxychlorzoxazone, 4′-hydroxymephenytoin, and hydroxytolbutamide were obtained from Ultrafine Chemical Company (Manchester, UK), and 7-hydroxycoumarin and 7-ethoxyresorufin were purchased from Sigma-Aldrich. HPLC-grade solvents were obtained from Rathburn Chemicals Ltd. (Walkerburn, UK). The laboratory water was purified trough a Milli-Q system (Millipore Corporation, Billerica, MA). Other chemicals used were mainly from Sigma-Aldrich and Boehringer Ingelheim GmbH (Ingelheim, Germany) and were of the highest purity available.
Human Liver Microsomes and cDNA-Expressed Human P450s. Human liver samples used in this study were obtained from the University Hospital of Oulu as surplus from kidney transplantation donors. The collection of surplus tissue was approved by the Ethics Committee of the Medical Faculty of the University of Oulu, Finland. The livers were transferred to ice immediately after the surgical excision, cut into pieces, snap-frozen in liquid nitrogen, and stored at -80°C until the microsomes were prepared by standard differential ultracentrifugation (Pelkonen et al., 1974). A weight-balanced microsomal pool of seven liver microsomal preparations that have been extensively characterized to be used for primary screening (sufficient model activities, no known polymorphisms, expected effects of model inhibitors, quantitation of P450s by Western blotting) was used. The final microsomal pellet was suspended in 100 mM phosphate buffer, pH 7.4. Protein content was determined by the method of Bradford (1976). Baculovirus insect cell-expressed human CYP2B6 was purchased from BD Biosciences Discovery Labware (Bedford, MA) and was used according to manufacturer's instructions.
Enzyme Assays. The incubation conditions and instrumentation used to assess the enzyme activities of CYP1A1/2 (ethoxyresorufin O-de-ethylation), CYP2A6 (coumarin 7-hydroxylation), CYP2C9 (tolbutamide hydroxylation), CYP2C19 (mephenytoin 4′-hydroxylation), CYP2D6 (dextromethorphan O-demethylation), CYP2E1 (chlorzoxazone 6-hydroxylation), CYP3A4/5 (midazolam 1′-hydroxylation), and multiple P450s (7-ethoxycoumarin O-de-ethylation) were described previously in detail by Taavitsainen et al. (2001). The bupropion hydroxylation assay for CYP2B6 was slightly modified from Faucette et al. (2000) and Hesse et al. (2000): incubation mixtures contained 2.0 mg of microsomal protein/ml, 50 μM bupropion, 0.1 M phosphate buffer (pH 7.4), 1 mM NADPH, and one of the studied inhibitors at five different concentrations in a total volume of 200 μl. Each reaction mixture was preincubated for 2 min at 37°C in a shaking incubator block (Eppendorf Thermomixer 5436; Eppendorf-5 Prime, Inc., Boulder, CO). The reaction was started by adding NADPH. In mechanism-based inhibition experiments, an inhibitor was incubated with NADPH for 15 min (preincubation) before the reaction was started by the addition of bupropion. Each reaction was terminated by adding 200 μl of ice-cold acetonitrile and subsequently cooled in an ice bath to precipitate the proteins. The mixture was vortex-mixed and spun at 10,000g for 15 min. Supernatants were collected and stored at -20°C until analyzed.
In Vitro Inhibition and Reference Inhibitors. In the primary screening of the IC50 values, the inhibitors were added in five different concentrations (final concentrations in the incubation mixture: 0.01, 0.1, 1, 10, 100, and 1000 μM; the highest concentration of xanthate C8 was 500 μM) into the incubation mixture in a small volume of an appropriate solvent. The solvents used were methanol (benzylisothiocyanate, deramciclane, ethynylestradiol, l-methamphetamine, ketoconazole, nicotine, orphenadrine, ospemifene, and tamoxifen), ethanol (quercetin), dimethyl sulfoxide (entacapone, furafylline, nitecapone, retinol, thioTEPA, and tolcapone), acetonitrile (furafylline and sulfaphenazole), and water (xanthate C8, diethyldithiocarbamate, methimazole, metyrapone, pyridine, quinidine, selegiline, ticlopidine, and tranylcypromine). The final amount of primary solvents in the incubation mixture was under 1% (v/v) for both controls and incubates containing inhibitors. The IC50 values for inhibitors were determined graphically by a linear regression analysis of the plot of the logarithm of inhibitor concentration versus percentage of activity remaining after inhibition using Microcal Origin, version 6.0 (OriginLab Corp., Northampton, MA). All datapoints represent the average of duplicate incubations. The enzyme activities in the presence of inhibitors were compared with the control incubations (incubations containing solvent but no inhibitor).
For determining the Ki values and mode of inhibition of ticlopidine and thioTEPA, three concentrations of bupropion (50, 100, and 200 μM, corresponding approximately to Km/2, Km, and 2 × Km) were incubated with a range of inhibitor concentrations in the presence of human liver microsomes or recombinant CYP2B6, as described above. Graphical analysis of data was performed according to Lineweaver-Burk, Dixon, Hofstee, and Hanes plots.
HPLC Analysis. For the HPLC method of the metabolites of bupropion, chlorzoxazone, dextromethorphan, rac-mephenytoin, midazolam, and tolbutamide, the peaks were separated and resolved at ambient temperature on a Symmetry C18 column (3.9 × 150 mm, 5-μm particle size; Waters, Milford, MA) preceded by a Lichospher 100 RP-18 guard column (4 × 4 mm; Merck, Darmstadt, Germany). For UV-HPLC analysis, samples were centrifuged at 10,000g for 15 min before injection into HPLC. The apparatus used was a Shimadzu VP series HPLC with autoinjector (Shimadzu, Kyoto, Japan). Mobile phases were pumped over a gradient at a flow rate of 1 ml/min. The injection volume was 20 μl. Concentrations of metabolites were calculated from peak height ratios on the basis of standard calibration curves of authentic metabolites. The variability between parallel incubates was less than 15% in all samples.
Results
An initial screening of altogether 30 compounds with respect to inhibition of CYP2B6-mediated bupropion hydroxylation indicated that ticlopidine, thioTEPA, metyrapone, xanthate C8, and benzylisothiocyanate are relatively potent CYP2B6 inhibitors (Fig. 1) with IC50 values of 0.32, 1.75, 4.14, 6.93, and 8.59 μM, respectively (Table 1). These chemicals were selected for more detailed studies. A number of other compounds tested exhibited modest inhibitory potency (IC50 values between 10 and 100 μM); these included retinol, deramciclane, troglitazone, and ethinylestradiol (Table 1). The screening set included also a number of P450-selective “diagnostic” inhibitors. It is worth noting that the CYP2A6 inhibitor tranylcypromine and the CYP3A4 inhibitor ketoconazole inhibited bupropion hydroxylation with IC50 values of 3.1 and 3.5 μM, respectively (Table 1). Of the other chemicals tested, diethyldithiocarbamate, entacapone, haloperidol, lidocaine, l-methamphetamine, melperone, methimazole, nicotine, nitecapone, orphenadrine, repaglinide, tamoxifen, tolcapone, and the P450-specific diagnostic inhibitors, furafylline, quercetin, sulfaphenazole, fluconazole, quinidine, and pyridine, showed no inhibition, even at the concentration level of 100 μM (data not shown).

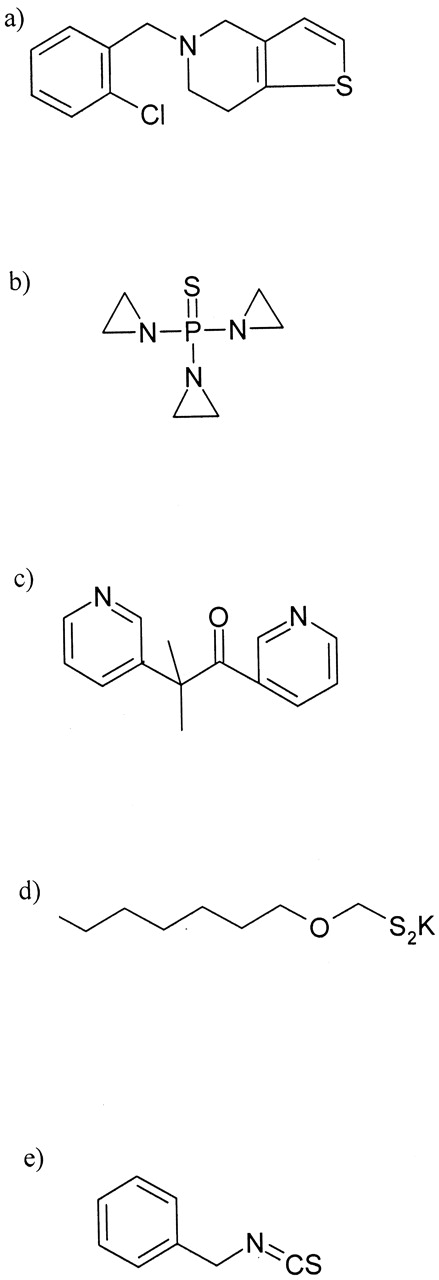
Chemical structures of ticlopidine (a), thioTEPA (b), metyrapone (c), xanthate C8 (d), and benzylisothiocyanate (e).
Inhibition of CYP2B6-catalyzed bupropion hydroxylation by various substances in human liver microsomes
The selected set of potent CYP2B6 inhibitors was studied for inhibitory potency toward other major P450-catalyzed activities (Table 2). The IC50 value of benzylisothiocyanate for CYP2B6 was 8.59 μM, but it also inhibited all the other activities studied except tolbutamide hydroxylation (CYP2C9) and chlorzoxazone 6-hydroxylation (CYP2E1). Thus, benzylisothiocyanate is rather unselective toward various P450 enzymes and unsuitable as a CYP2B6 selective inhibitor.
IC50 values (μM) of ticlopidine, thioTEPA, metyrapone, xanthate C8, and benzylisothiocyanate on P450-specific model activities in human liver microsomes Mechanism-based inhibition experiments (15-min preincubation) for CYP2B6 are also presented. Substrate concentrations in parentheses.
The IC50 value of xanthate C8 was the lowest, 1.8 μM, against chlorzoxazone 6-hydroxylation (CYP2E1) (Table 2). Inhibition of CYP2A6 was also seen with xanthate C8, and two other enzymes, CYP2B6 and 2C19, were inhibited to the same extent. The IC50 values of these three enzymes were only a few times higher than that seen with the CYP2E1 model reaction. The IC50 values of CYP1A2, 2C9, 2D6, and 3A4 were many times higher compared with the IC50 value of xanthate C8 in chlorzoxazone 6-hydroxylation. Xanthate C8 thus displays considerable unselectivity with respect to P450 enzymes.
Metyrapone, a classic CYP2B inhibitor in experimental animals, potently inhibited dextromethorphan O-demethylation (CYP2D6) in human liver microsomes, as shown by an IC50 value of 1.06 μM for incubations with 100 μM dextromethorphan (Table 2). Compared with CYP2B6, the IC50 value of metyrapone on CYP2D6-catalyzed reaction was over 2.5-fold smaller. The inhibitions of CYP1A2, CYP2A6, CYP2C9, CYP2C19, and CYP2E1 were weaker, as indicated by IC50 values of >10 μM. Thus, metyrapone is not selective enough as a CYP2B6 inhibitor.
In addition to CYP2B6, ticlopidine was also shown to have a relatively potent inhibitory effect on mephenytoin 4′-hydroxylase (CYP2C19) activity and dextromethorphan O-demethylase (CYP2D6) activity, the IC50 values being 2.7 and 4.4 μM, respectively. The inhibition of CYP2A6-, 2C9-, 2E1-, and 3A4-associated activities by ticlopidine were minimal, as indicated by IC50 values >500 μM (Table 2). At a 1 μM concentration of ticlopidine, a complete inhibition of bupropion hydroxylation activity was observed. In comparison, only a minimal inhibition of the other P450-related activities was seen at this concentration level. Essentially similar findings were obtained with recombinant expressed P450 enzymes (Fig. 2). Although some effect on ethoxyresorufin O-deethylase (CYP1A2) and mephenytoin 4-hydroxylase (CYP2C19) activities was observed, the extent of ticlopidine inhibition at 1 μM was, on average, only about 13%.

Effect of ticlopidine at the “most selective” concentration (1 μM) on P450 enzyme activities in human liver microsomes (HLM) and recombinant enzymes.
Each data point represents the mean of duplicate determinations.
Ticlopidine showed no evidence of a suicidal inhibition mode toward 7-ethoxycoumarin O-de-ethylation, the IC50 values being 24.5 μM for 2-min and 33.1 μM for 15-min preincubation (data not demonstrated). However, when ticlopidine was preincubated with NADPH for 15 min before bupropion was added, there was a severalfold decrease of the IC50 value to 0.08 μM (Table 2), indicating a definite component of mechanism-based inhibition mode in ticlopidine effect. This finding is in accordance with the mode of inhibition of bupropion hydroxylation by ticlopidine (see below). None of the other inhibitors displayed a similar degree of reduction of IC50 values. Instead, the IC50 value of xanthate C8 after the 15-min preincubation was several times higher than without preincubation.
ThioTEPA displayed the best selectivity toward various P450-associated model activities; an IC50 value of 1.75 μM for CYP2B6 and an IC50 of 256 μM for CYP2A6. All the other activities were inhibited even less potently (CYP3A4) or not at all (all the other P450 isoforms studied).
The mode of inhibition of bupropion hydroxylation by ticlopidine in human liver microsomes and recombinant CYP2B6 was found to be of mixed type with the Ki value of 0.2 μM (Fig. 3a). ThioTEPA inhibited bupropion hydroxylation competitively with a Ki value of 2.8 μM in both human liver microsomes and recombinant P450s (Fig. 3b). The calculated theoretical Ki values derived from the Tornheim equation (Tornheim, 1994) were 0.2 μM for ticlopidine and 1.1 μM for thioTEPA.

A representative Dixon plot for the inhibition of bupropion hydroxylation by ticlopidine (a) and thioTEPA (b) in human liver microsomes.
The inhibitor concentrations were 0.1, 0.25, 0.5, and 1 μM for ticlopidine and 0.5, 1, 2.5, and 5 μM for thioTEPA. Each data point represents the mean of duplicate determinations.
Discussion
In this study, the goal was to find and characterize a selective and potent chemical inhibitor for CYP2B6 enzyme activity in human liver microsomes. After a screening of >30 compounds, and a more detailed investigation of five selected compounds, ticlopidine, thio-TEPA, metyrapone, xanthate C8, and benzylisothiocyanate, two useful in vitro inhibitors, ticlopidine and thioTEPA, were found to be potent and relatively selective inhibitors of CYP2B6.
The apparent IC50 values of ticlopidine for each enzyme in human liver microsomes showed that it is a relatively selective inhibitor of CYP2B6. Previously published reports (Ko et al., 2000; Giancarlo et al., 2001; Ha-Duong et al., 2001) have shown ticlopidine to be a selective mechanism-based inhibitor of human cytochrome P450 2C19. A recent study from Richter et al. (2004) demonstrated ticlopidine as a mechanism-based inhibitor of CYP2B6, as well as the inhibitory effect against CYP2C19. Our present results are in good accordance with those from Richter et al. Also in this study, a relatively potent inhibitory effect against CYP2C19 activity was seen, the IC50 being 2.7 μM. This is 8.4-fold above the IC50 value of ticlopidine toward CYP2B6-mediated bupropion hydroxylation. Although some inhibition of CYP1A2 and CYP2D6 was observed, their IC50 values were at least 1 order of magnitude higher than those of CYP2B6 and CYP2C19. Inhibitory effects of ticlopidine on other P450 forms were negligible. If the calculated Ki values of ticlopidine in other P450-specific activities are divided by the Ki value of ticlopidine in bupropion hydroxylation, a rank of inhibitory potencies (5.05 for CYP2C19, 7.35 for CYP2D6, 18.9 for CYP1A2, 54.6 for CYP2A6, 1071 for CYP2E1, and no inhibition for CYP2C9 and CYP3A4) shows that ticlopidine is over 5 times more potent an inhibitor of CYP2B6 than any of the other enzymes studied here. On the other hand, based on these results, ticlopidine is not a good reference inhibitor for CYP2C19, because it inhibits CYP2B6 much more potently.
These data clearly demonstrate that ticlopidine is able to inhibit human liver CYP2B6 in vitro and may contribute to clinical interactions with substrates of this P450 isoform. Bupropion is used as an antismoking agent, but it also has antidepressive properties. The risk of drug interactions or central nervous system toxicity associated with bupropion may be of clinical importance and correlates with high bupropion or metabolite levels. Since ticlopidine has been approved as an antiplatelet drug for patients at high risk of vascular disease, its use is expected to increase (Hankey et al., 2000). Ticlopidine is a thienopyridine chemically related to the other member of the group, clopidogrel (Algra and van Gijn, 2000). Because of the similarities in the structures of these two drugs, it would be interesting to study the inhibitory potency of both drugs toward CYP2B6 and other P450 activities in vivo.
ThioTEPA was included in this study because of the results of Rae et al. (2002). They demonstrated that thioTEPA is a potent and selective CYP2B6 inhibitor with an IC50 value near 5 μM using mephenytoin biotransformation to nirvanol as a CYP2B6 model reaction. The type of inhibition of CYP2B6 by thioTEPA was found to be noncompetitive in both human liver microsomes and recombinantly expressed CYP2B6 with Ki values of approximately 5 μM. In our present studies with bupropion hydroxylation, the inhibitory effect of thioTEPA toward CYP2B6 was found to be competitive, with Ki value of 2.8 and IC50 value of 1.75 μM.
Metyrapone has been proposed to be a selective chemical inhibitor of CYP2B subfamily in rat liver microsomes (Lambard et al., 1991). In our previous studies with murine and human livers (Mäenpää et al., 1991), metyrapone inhibited strongly CYP2A6 associated coumarin 7-hydroxylation in mouse liver microsomes but not in microsomes from human origin; in human liver microsomes, metyrapone inhibited mainly CYP3A4-mediated testosterone 6β-hydroxylation. In the present study, inhibition of CYP3A4 and unaffected CYP2A6 activity were consistently observed. In addition, metyrapone inhibited CYP2D6. Both CYP2D6 and CYP3A4 enzyme activities were inhibited even more potently than CYP2B6.
Of the other inhibitors studied more thoroughly here, neither xanthate C8 nor benzylisothiocyanate is a selective inhibitor of CYP2B6. Xanthates have been previously reported to be selective mechanism-based inactivators of CYP2B6 (Kent et al., 1999; Yanev et al., 1999). The present study shows that xanthate C8 is not a good reference inhibitor for CYP2B6 because it inhibits many other P450-specific enzyme activities (CYP2A6, 2C19, and 2E1) more potently. The IC50 value of xanthate C8 against chlorzoxazone 6-hydroxylation was interestingly even lower than the IC50 value of pyridine, the reference inhibitor used in CYP2E1 studies. Benzylisothiocyanate inhibited CYP1A2, CYP2D6, and CYP2C19 rather potently (IC50 values: 5.6, 4.8, and 7.2 μM, respectively), and CYP2A6 and CYP3A4 moderately. It is clear that these two substances are not suitable for use as selective CYP2B6 inhibitors.
CYP2A6 inhibitor tranylcypromine (Taavitsainen et al., 2001) and the CYP3A4 inhibitor ketoconazole (Baldwin et al., 1995; Newton et al., 1995) also inhibited CYP2B6 enzyme quite potently. However, the IC50 values against CYP2B6 are still 10 to 50 times greater than those we have obtained previously against CYP2A6 (0.39 μM for tranylcypromine) and 3A4 (0.065 μM for ketoconazole). This information may be worthwhile to note when these inhibitors are used as diagnostic inhibitors or particular enzyme activities need to be suppressed.
It has to be acknowledged that selectivity applies to only the enzymes tested. Probably the most important enzyme, which was not studied here, is CYP2C8. Rae et al. (2002) measured the effect of thioTEPA on CYP2C8-associated tolbutamide 4-methylhydroxylation and found no inhibition (Rae et al., 2002). We have measured, as a part of a cocktail approach (M. Turpeinen, J. Uusitalo, J. Jalonen, and O. Pelkonen, unpublished data), the effect of ticlopidine on amodiaquine N-desethylation, another CYP2C8-associated activity (Li et al., 2002), and found that the IC50 value was higher than 100 μM. Thus, it seems that at least CYP2C8 is not inhibited by ticlopidine or thioTEPA.
In conclusion, both ticlopidine and thioTEPA are highly effective and potent inhibitors of CYP2B6. Ticlopidine, at the concentration that inhibits the CYP2B6 activity completely (1 μM), has less than 15% inhibitory effect on the activities of other major human hepatic P450s. Although ticlopidine is 10-fold more potent than thioTEPA and even more potent in a mechanism-based inhibition mode, thio-TEPA was found to be about 2 orders of magnitude more selective than ticlopidine. These observations suggest that both ticlopidine and thioTEPA would be useful as probe substances in testing CYP2B6 activity in vitro. If high selectivity is needed, thioTEPA is the drug of choice. If high potency of in vivo application is needed, ticlopidine is a useful and moderately selective agent.
Acknowledgments
The excellent technical assistance of Anne Vuollo, Esa Kerttula, Ritva Tauriainen, and Päivi Tyni is greatly acknowledged. This study belongs to the Finnish National Research Programme “Drug2000” (subsection “screening methods”).
Footnotes
-
↵2 Abbreviations used are: P450, cytochrome P450; thioTEPA, triethylenethiophosphoramide; HPLC, high-performance liquid chromatography.
-
This work was funded by grants from National Technology Agency of Finland (TEKES).
-
↵1 Current address: Orion Pharma, Department of Non-clinical Pharmacokinetics, P.O. Box 425, 20101 Turku, Finland.
- Received November 5, 2003.
- Accepted February 23, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}