


Abstract
Capecitabine, an anticancer prodrug, is thought to be biotransformed into active 5-fluorouracil (5-FU) by three enzymes. After oral administration, capecitabine is first metabolized to 5′-deoxy-5-fluorocytidine (5′-DFCR) by carboxylesterase (CES), then 5′-DFCR is converted to 5′-deoxy-5-fluorouridine (5′-DFUR) by cytidine deaminase. 5′-DFUR is activated to 5-FU by thymidine phosphorylase. Although high activities of drug metabolizing enzymes are expressed in human liver, the involvement of the liver in capecitabine metabolism is not fully understood. In this study, the metabolism of capecitabine in human liver was investigated in vitro. 5′-DFCR, 5′-DFUR, and 5-FU formation from capecitabine were investigated in human liver S9, microsomes, and cytosol in the presence of the inhibitor of dihydropyrimidine dehydrogenase, 5-chloro-2,4-dihydroxypyridine. 5′-DFCR, 5′-DFUR, and 5-FU were formed from capecitabine in cytosol and in the combination of microsomes and cytosol. Only 5′-DFCR formation was detected in microsomes. The apparent Km and Vmax values of 5-FU formation catalyzed by cytosol alone and in combination with microsomes were 8.1 mM and 106.5 pmol/min/mg protein, and 4.0 mM and 64.0 pmol/min/mg protein, respectively. The interindividual variability in 5′-DFCR formation in microsomes and cytosol among 14 human liver samples was 8.3- and 12.3-fold, respectively. Capecitabine seems to be metabolized to 5-FU in human liver. 5′-DFCR formation was exhibited in cytosol with large interindividual variability, although CES is located in microsomes in human liver. In the present study, it has been clarified that the cytosolic enzyme would be important in 5′-DFCR formation, as is CES.
5-Fluorouracil (5-FU) is one of the most widely used anticancer agents in the chemotherapy of solid tumors. However, the efficacy of 5-FU is limited due to its rapid degradation into dihydro-5-fluorouracil when catalyzed by dihydropyrimidine dehydrogenase (DPD), and toxicity is observed because of the lack of selectivity toward tumors (Ishikawa et al., 1998b). Prodrugs of 5-FU have been developed to improve the safety and efficacy profile of 5-FU. For example, tegafur [5-fluoro-1-[(RS)-tetrahydrofuran-2-yl]pyrimidine-2,4(1H,3H)-dione] is an extended-release drug that maintains the effective 5-FU concentration over a long period. Doxifluridine (5′-deoxy-5-fluorouridine; 5′-DFUR) has a higher selectivity toward tumors than 5-FU. Oral administration requires higher dosages than intravenous injection due to first pass effects. Adverse effects such as diarrhea have been reported following the administration of tegafur or doxifluridine (Ohta et al., 1980; Ninomiya et al., 1990).
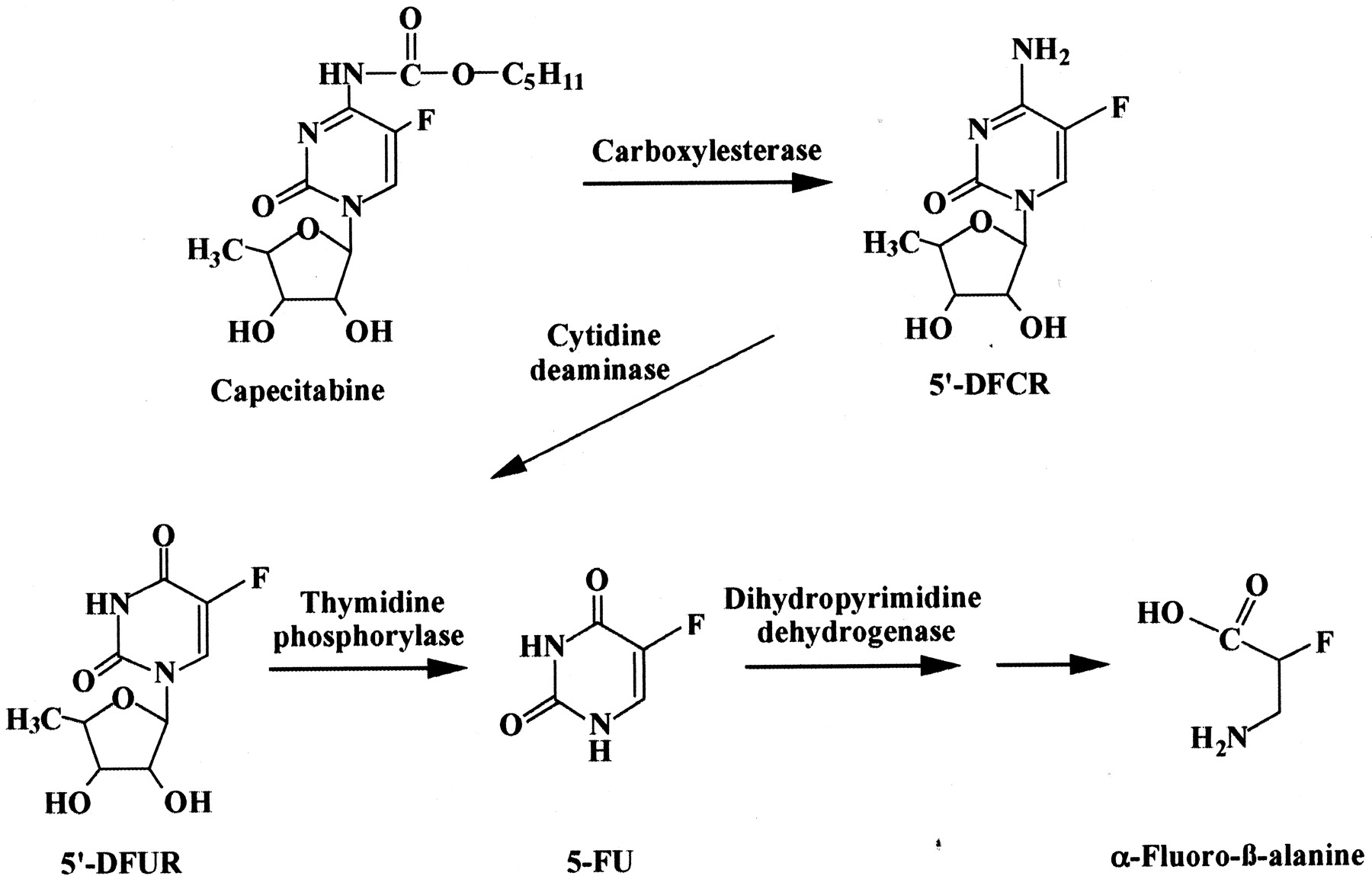
Capecitabine (N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine) was designed to reduce the adverse effects and improve the selectivity toward tumors. Capecitabine is a novel oral fluoropyrimidine carbamate aimed at preferential conversion to 5-FU within the tumors and has been clinically used in many countries, including Japan and the United States, and in Europe. It is considered to be bioactivated into 5-FU by three enzymes (Fig. 1): carboxylesterase (CES), cytidine deaminase (CDA), and thymidine phosphorylase (TP). In humans, CES is mainly expressed in the liver and intestine (Junge et al., 1974; Inoue et al., 1979). CDA is found in most tissues, including the liver and in tumor tissues (Camiener and Smith, 1965). TP is expressed in many tissues throughout the body (Yoshimura et al., 1990). Some human carcinomas express TP at higher levels than the surrounding normal tissues (Kono et al., 1981; Nishida et al., 1996). It has been reported that the concentration of 5-FU is higher in tumor tissues than the surrounding normal tissues, muscle, or plasma after oral administration of capecitabine (Ishikawa et al., 1998b; Schüller et al., 2000). It has been proposed that the higher sensitivity of capecitabine is mainly caused by TP localization, although TP is highly expressed in the liver. However, the hepatic metabolism of capecitabine is not fully understood, especially the involvement of CES.

Proposed metabolic pathway of capecitabine in humans.
The purpose of this study is to clarify the in vitro metabolism of capecitabine in human liver. Regarding prodrugs, the results would provide valuable information for elucidating its metabolic fate in chemotherapy.
Materials and Methods
Chemicals. Capecitabine, 5′-deoxy-5-fluorocytidine (5′-DFCR), 5′-DFUR, 5-FU, 5-chlorouracil, 5-chloro-2,4-dihydroxypyridine (CDHP), and 5-chloro-6-(2-iminopyrrolidin-1-yl)methyl-2,4-(1H,3H)-pyrimidinedione (TPI) were kindly provided by Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan). Diisopropylfluorophosphate (DFP) was purchased from Wako Pure Chemicals (Osaka, Japan). Bis(p-nitrophenyl)phosphate (BNPP) and tetrahydrouridine (THU) were obtained from Sigma-Aldrich (St. Louis, MO) and EMD Biosciences (San Diego, CA), respectively. Other chemicals used in this study were of the highest quality commercially available.
Enzyme Sources. For the kinetic and inhibition study, pooled human liver microsomes, pooled human liver cytosol, and pooled human liver S9 were obtained from BD Gentest (Woburn, MA). Human liver cytosol and S9 were dialyzed using dialysis membrane size 8 (Wako Pure Chemicals) against 300 volumes of 20 mM potassium phosphate buffer (pH 7.4) containing 1 mM 2-mercaptoethanol according to the methods described previously (Ishikawa et al., 1998a) and were used as human liver cytosol and S9 in the following experiments. For the investigation of interindividual variability on 5′-DFCR formation, human liver microsomes and human liver cytosol from single donor were purchased from BD Gentest, except K19, K20, and K41. Human liver samples from three Japanese individuals (K19, K20, and K41) were obtained at autopsy. The use of the human livers was approved by the Institutional Committee of Dokkyo University School of Medicine (Tochigi, Japan). Liver tissues were rapidly frozen in liquid nitrogen immediately after excision and stored at -80°C. Liver tissues were homogenized in three volumes of 0.1 M Tris-HCl buffer (pH 7.4) containing 1 mM EDTA and 0.1 M KCl, and the homogenate was centrifuged at 9,000g for 15 min. The supernatant was further centrifuged at 105,000g for 90 min to prepare the microsomes. The human liver microsomes were prepared in 10 mM Tris-HCl buffer (pH 7.4) containing 0.1 mM EDTA and 20% (v/v) glycerol as described previously (Yamazaki et al., 1999). The 105,000g supernatant was dialyzed as described above and used as human liver cytosol. The protein concentration was measured by the method of Lowry et al. (1951) using bovine serum albumin as a standard. The recombinant human carboxylesterases HU1 and HU3 were prepared as described previously (Furihata et al., 2003).
Characterization of Capecitabine Metabolism in Human Liver in Vitro. Activities for 5′-DFCR, 5′-DFUR, and 5-FU formations were determined as follows. A typical standard reaction mixture (total volume of 0.25 ml) consisted of the enzyme source, 30 mM Tris-HCl buffer (pH 7.4) containing 92.4 mM KCl and 1.2 mM EDTA, 2.5 mM nicotinamide adenine dinucleotide phosphate (reduced form), 100 μM CDHP, and 100 μM capecitabine. In all reaction mixtures, CDHP, an inhibitor of DPD (Tatsumi et al., 1987), was added. Capecitabine was dissolved in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the reaction mixture was <0.75%. The reaction was initiated by the addition of capecitabine after a 2-min preincubation at 37°C. After incubation for 30 min at 37°C, the reaction was terminated by adding 1.5 ml of ethyl acetate and 10 μl of 0.5 M HCl. 5-Chlorouracil was added as an internal standard. The reaction mixture was extracted twice with 1.5 ml of ethyl acetate. After centrifugation at 650g for 10 min, the organic phase was evaporated to dryness under a gentle N2 stream. The residue was dissolved in 100 μl of distilled water. The product formations were determined by high-performance liquid chromatography (HPLC). HPLC analysis was performed using an LC-6A pump (Shimadzu, Kyoto, Japan), SPD-6AV UV detector (Shimadzu), SIL-9A autosampler (Shimadzu), SLC-6B system controller (Shimadzu), Chromatopac C-R7A plus integrator (Shimadzu), noisebase clean Uni-3 (Union Co., Ltd., Gunma, Japan), and a CO-965 column oven (Jasco, Tokyo, Japan) equipped with a C30 analytical column (Develosil C30-UG-5, 150 × 4.6 mm; Nomura Chemical Co. Ltd., Aichi, Japan). The flow rate was 1.0 ml/min, and the column temperature was 30°C. The mobile phases were solvent A (10 mM sodium phosphate buffer, pH 4.8) and solvent B (80% methanol). A typical condition for the elution was as follows: 97% A (0–9 min), 97–0% A (9–35 min), 0% A (35–40 min), and 0–97% A (40–42 min). A linear gradient was used for all solvent changes. The eluent was monitored at 265 nm (5′-DFCR, 5′-DFUR, and 5-FU) and 315 nm (capecitabine). The 5′-DFCR formation was quantified using a standard curve of 5′-DFUR because we could not obtain authentic 5′-DFCR.
In this study, pooled human liver S9, pooled human liver microsomes, pooled human liver cytosol, HU1, and HU3 were used as the enzyme sources. The protein concentrations of S9, microsomes, cytosol, the combination of microsomes and cytosol, HU1, and HU3 were 1.5 mg/ml, 0.2 mg/ml, 1.0 mg/ml, 1.2 mg/ml, 0.02 mg/ml, and 0.02 mg/ml, respectively. All data were analyzed using the mean of duplicate determinations.
Inhibition Studies. For inhibition studies of BNPP, DFP, THU, and TPI, the protein concentrations of microsomes, cytosol, and the combination of microsomes and cytosol were 0.5 mg/ml, 2.5 mg/ml, and 3.0 mg/ml, respectively. BNPP and DFP were used as inhibitors of CES (Brandt et al., 1980). THU and TPI were used as inhibitors of CDA (Wentworth and Wolfenden, 1975) and TP (Fukushima et al., 2000), respectively. BNPP, DFP, THU, and TPI were added to the reaction mixtures at 0.1 to 10 μM, 0.01 to 1 μM, 0.04 to 4 μM, and 5 to 100 nM, respectively. For inhibition studies on 5′-DFCR formation, BNPP and DFP ranged from 0.1 to 10 μM and 5 nM to 1 μM. BNPP, CDHP, THU, and TPI were dissolved in water, and DFP was dissolved in methanol. The final concentration of organic solvent in the reaction mixture was <1.0%.
Kinetic Analyses of 5′-DFCR and 5-FU Formations. The kinetic analyses of 5′-DFCR and 5-FU formation from capecitabine were performed at the range of 0.05 to 3 mM. For the measurement of 5′-DFCR formation, THU (500 μM) and TPI (0.1 μM) were added to the reaction mixtures. Kinetic parameters were calculated from the fitting curves by nonlinear regression.
Results
Analyses for 5′-DFCR, 5′-DFUR, and 5-FU Formation Catalyzed by Human Liver. 5′-DFCR, 5′-DFUR, and 5-FU formation from capecitabine were determined by HPLC. Typical HPLC chromatograms are shown in Fig. 2. The retention time of 5′-DFCR was confirmed using the incubation product of CES from porcine liver (Sigma-Aldrich) and capecitabine. Since the trace peaks corresponding to 5′-DFCR and 5′-DFUR were observed when the reaction mixture was not incubated, the background levels were subtracted in the calculation of the enzymatic activities for 5′-DFCR and 5′-DFUR formations at each substrate concentration.

Representative HPLC chromatograms of capecitabine metabolites catalyzed by the combination of human liver microsomes and cytosol. A, authentic standards: 1) 5-FU; 2) 5-chlorouracil (internal standard); 3) 5′-DFCR; 4) 5′-DFUR; and 5) capecitabine. B, capecitabine (100 μM) was incubated at 37°C for 30 min with the combination of human liver microsomes (0.2 mg/ml) and cytosol (1.0 mg/ml) in the presence of CDHP (100 μM). Capecitabine was also detected at 315 nm.
Characterization of Capecitabine Metabolism in Human Liver in Vitro. To clarify the in vitro metabolism of capecitabine in human liver, 5′-DFCR, 5′-DFUR, and 5-FU formation from capecitabine in liver S9 was investigated (Fig. 3). 5′-DFCR, 5′-DFUR, and 5-FU formation was increased in an incubation time-dependent manner.

Changes of metabolic profiles of capecitabine in human liver S9. Capecitabine (100 μM) was incubated with S9 (1.5 mg/ml) obtained from BD Gentest for 0 to 120 min. Each data point represents the mean of duplicate determinations.
Inhibition Studies on 5′-DFCR, 5′-DFUR, and 5-FU Formation. Inhibitory effects of BNPP, DFP, THU, and TPI on 5′-DFCR, 5′-DFUR, and 5-FU formation from capecitabine are shown in Table 1. Since liver S9 contained approximately 5-fold more cytosolic protein than the microsomal protein, cytosol was used at a protein concentration 5 times higher than that of the microsomes in the present study. 5′-DFCR, 5′-DFUR, and 5-FU formation in the combination of microsomes and cytosol was almost the same as that in S9 (data not shown). 5′-DFCR formation activity was inhibited by BNPP and DFP in a concentration-dependent manner. At 1 μM THU, 5′-DFUR formation disappeared, but 5-FU formation was detected. 5-FU formation was not detected at 400 μM THU (data not shown). 5-FU formation was inhibited completely at 0.1 μM TPI. As shown in Table 2, the apparent IC50 values of BNPP and DFP for 5′-DFCR formation were 740 and 154 nM, respectively. The apparent IC50 values of THU for 5′-DFUR formation and TPI for 5-FU formation were 55 and 10 nM, respectively.
Effects of chemical inhibitors on 5′-DFCR, 5′-DFUR, and 5-FU formation in the combination of human liver microsomes and cytosol Capecitabine (100 μM) was incubated with the combination of microsomes (0.5 mg/ml) and cytosol (2.5 mg/ml) obtained from BD Gentest in the presence or absence of chemical inhibitors. BNPP, DFP, THU, and TPI concentrations ranged from 0.1 to 10 μM, 0.01 to 1 μM, 0.04 to 4 μM, and 5 to 100 nM, respectively. Each data represents the mean of duplicate determinations. Values in parentheses indicate the percentages of the control activities.
Apparent IC50 values of BNPP, DFP, THU, and TPI on 5′-DFCR, 5′-DFUR, and 5-FU formations Capecitabine (0.05—3 mM) was incubated with the combination of microsomes (0.5 mg/ml) and cytosol (2.5 mg/ml) obtained from BD Gentest. BNPP, DFP, THU, and TPI concentrations ranged from 0.1 to 10 μM, 0.01 to 1 μM, 0.04 to 4 μM, and 5 to 100 nM, respectively.
Kinetic Analysis of 5-FU Formation. The kinetic parameters of 5-FU formation from capecitabine are shown in Table 3. 5-FU formation was not detected in microsomes but was detected in cytosol. The apparent Km and Vmax values of 5-FU formation in the combination and in cytosol alone were 4.0 mM and 64.0 pmol/min/mg protein and 8.1 mM and 106.5 pmol/min/mg protein, respectively.
Kinetic parameters of 5-FU formation from capecitabine in the combination of human liver microsomes and cytosol, microsomes, and cytosol Capecitabine (0.05—3 mM) was incubated with the combination of microsomes and cytosol (1.2 mg/ml), microsomes (0.2 mg/ml), and cytosol (1.0 mg/ml) obtained from BD Gentest for 30 min in the presence of CDHP. The kinetic parameters were calculated from the fitted curves by nonlinear regression. The detection limit of 5-FU was 1.0 pmol.
Kinetic Analysis of 5′-DFCR Formation. To clarify the involvement of cytosolic enzyme in the capecitabine metabolism, we focused on the pathway of the 5′-DFCR formation from capecitabine. The calculated kinetic parameters of 5′-DFCR formation from capecitabine are shown in Table 4. The apparent Km values of 5′-DFCR formation in the combination of microsomes and cytosol, microsomes, and cytosol were 3.8, 3.4, and 1.5 mM, respectively. The apparent Vmax values in the combination, microsomes, and cytosol were 136.1, 420.2, and 40.1 pmol/min/mg protein, respectively. The clearance in microsomes was approximately 5-fold higher than that in cytosol.
Kinetic parameters of 5′-DFCR formation from capecitabine in the combination of human liver microsomes and cytosol, microsomes, and cytosol Capecitabine (0.05—3 mM) was incubated with the combination of microsomes and cytosol (1.2 mg/ml), microsomes (0.2 mg/ml), and cytosol (1.0 mg/ml) obtained from BD Gentest for 30 min in the presence of THU (500 μM), TPI (0.1 μM), and CDHP (100 μM). The kinetic parameters were calculated from the fitted curves by nonlinear regression.
Inhibition Study of 5′-DFCR Formation. The inhibitory effects of BNPP and DFP on 5′-DFCR formation from capecitabine were investigated in the presence of CDA, TP, and DPD inhibitors (Table 5). 5′-DFCR formation catalyzed by the combination of microsomes and cytosol, microsomes, and cytosol were inhibited by both BNPP and DFP in a concentration-dependent manner. The IC50 value of BNPP in microsomes was higher than that in the combination and in cytosol.
Effects of CES inhibitors on 5′-DFCR formation in the combination of human liver microsomes and cytosol, microsomes, and cytosol Capecitabine (100 μM) was incubated with the combination of microsomes (0.5 mg/ml) and cytosol (2.5 mg/ml), microsomes (0.5 mg/ml), and cytosol (2.5 mg/ml) obtained from BD Gentest in the presence or absence of the CES inhibitor. BNPP and DFP concentrations ranged from 0.1 to 10 μM and 5 nM to 1.0 μM, respectively. The incubation contained the inhibitor mixture of 500 μM THU, 0.1 μM TPI, and 100 μM CDHP. Each data point represents the mean of duplicate determinations. Values in parentheses indicate the percentages of the control activities.
Interindividual Variability in Microsomal and Cytosolic 5′-DFCR Formation. To investigate the interindividual variability in microsomal and cytosolic 5′-DFCR formation, 14 samples of human liver microsomes and cytosol were examined at 100 μM capecitabine (Fig. 4). In microsomes, HG30 showed the highest activity (34.8 pmol/min/mg protein), and HG06 showed the lowest activity (4.2 pmol/min/mg protein). On the other hand, in cytosol, K19 showed the highest activity (9.8 pmol/min/mg protein), and HG112 showed the lowest activity (0.8 pmol/min/mg protein). The interindividual variability in 5′-DFCR formation in microsomes and cytosol among the 14 samples was 8.3- and 12.3-fold, respectively. The 5′-DFCR formation ratios (cytosol/microsomes) ranged from 0.1 to 2.1.

Interindividual variability in 5′-DFCR formation from capecitabine in human liver microsomes or cytosol. Capecitabine (100 μM) was incubated with microsomes (0.5 mg/ml) or cytosol (2.5 mg/ml) for 30 min in the presence of THU (500 μM), TPI (0.1 μM), and CDHP (100 μM). Except for K19, K20, and K41, both microsomes and cytosol were purchased from BD Gentest. Each column represents the means of duplicate determinations.
5′-DFCR Formation in CES Isoforms HU1 and HU3. 5′-DFCR formation in the human recombinant CESs, HU1 and HU3, was examined. 5′-DFCR formation in HU1 and HU3 was 275.2 and 9.4 pmol/min/mg protein at 100 μM capecitabine, respectively.
Discussion
Capecitabine is a novel fluoropyrimidine carbamate designed as an orally administered prodrug of 5-FU. Capecitabine seems to be sequentially metabolized by CES, CDA, and TP (Miwa et al., 1998). It is commonly considered that the preferential activation of capecitabine in tumors is caused by the tissue distribution of the key metabolic enzymes (Ishikawa et al., 1998b; Miwa et al., 1998; Schüller et al., 2000). In general, most drug metabolizing enzymes exhibit high activity in the liver; however, the hepatic metabolism of capecitabine is not fully understood, especially the catalysis by CES. In the present study, we investigated the metabolism of capecitabine in human liver.
Capecitabine was metabolized to 5-FU in a time-dependent manner in human liver S9 in the presence of CDHP (Fig. 3). This result means that capecitabine is possibly metabolized to the active metabolite in the human liver. On the other hand, it was demonstrated that capecitabine was metabolized to the final metabolite (α-fluoro-β-alanine; FBAL) in human liver S9 in the absence of CDHP (data not shown); however, FBAL formation in S9 was scarcely detected after a 90-min incubation. The amount of FBAL was quite small compared with that of 5-FU. The reason for this phenomenon may be that dihydropyrimidine dehydrogenase is a rate-limiting enzyme of 5-FU metabolism (Harris et al., 1998). Since the 5-FU concentration in human liver is not known, further investigation is needed to clarify the hepatic safety and efficacy of capecitabine.
The first step of capecitabine metabolism is thought to be its catalysis by CES. It has been determined that capecitabine is metabolized to 5′-DFCR in human liver, not in intestine (Shimma et al., 2000). The 5′-DFCR formation in various tumor tissues was lower than that in liver (Miwa et al., 1998). Therefore, the conversion from capecitabine to 5′-DFCR in the liver should be considered. The 5-FU formation in the liver was inhibited by BNPP and DFP (Table 1). It is recognized that DFP is a general inhibitor of the esterases and that BNPP is a more selective inhibitor of CES (Brandt et al., 1980). In the present study, the inhibitory potencies to 5-FU formation by BNPP and DFP were almost the same, suggesting that CES is involved in the metabolism of capecitabine in the liver. The human CES isoforms such as HU1, HU2, and HU3 have been purified (Satoh and Hosokawa, 1995). HU1 and HU3 would be predominantly expressed in human liver and intestine, respectively (Hosokawa et al., 1995). According to our investigation of the 5′-DFCR formation in recombinant human HU1 and HU3, the activity at 100 μM capecitabine in HU1 was approximately 30-fold higher than that in HU3. Thus, as suggested by Shimma et al. (2000), the first step of capecitabine metabolism will be carried out mainly in the liver.
Interestingly, it was shown that capecitabine was converted to 5-FU in cytosol (Table 3), although CES is not expressed in the cytosolic fraction in human liver (Satoh and Hosokawa, 1998). 5′-DFCR formation was observed in cytosol as well as microsomes in the presence of THU, TPI, and CDHP (Table 4). The apparent intrinsic clearance for 5′-DFCR formation in microsomes was approximately 5-fold higher than that in cytosol. Since liver S9 contains 5-fold more cytosolic protein than the microsomal protein, it is suggested that the contribution of cytosol on 5′-DFCR formation was almost the same as that of microsomes. We observed large interindividual variability in the 5′-DFCR formation in microsomes (8.3-fold) and cytosol (12.3-fold) in 14 human livers (Fig. 4). It has been reported that there was large interindividual variability in microsomal carboxylesterase activities and the expression levels in 12 human livers (Hosokawa et al., 1995). The carboxylesterase activities ranged from 5.3 (phenacetin) to 44.7 (p-nitrophenylpropionate) using 10 substrates. Since 5′-DFCR formation in microsomes would be catalyzed by CES in the present study, this is consistent with Hosokawa et al. (1995). It is noteworthy that the cytosolic enzyme also exhibited large interindividual variability in 5′-DFCR formation. In the present study, 5′-DFCR formation in cytosol was inhibited by BNPP and DFP (Table 5), indicating that the enzyme in cytosol has similar characteristics to those of CES. Although CES2 protein expression in human liver cytosol was suggested by Western blot analysis, there was no correlation between cytosolic CES activity and microsomal CES activity and between cytosolic CES activity and CES2 protein concentration (Xu et al., 2002). It was surmised that this cytosolic enzyme might be CES; therefore, the detailed investigation of this cytosolic enzyme is underway in our laboratory.
The conversion to 5′-DFUR from 5′-DFCR is thought to be catalyzed by CDA (Miwa et al., 1998). CDA is found in most human tissues, including the liver and tumors (Camiener and Smith, 1965). 5′-DFUR formation from 5′-DFCR in human liver is higher than that in various tumor tissues (Miwa et al., 1998). Thus, it is expected that 5′-DFCR is metabolized to 5′-DFUR mainly in the liver. It was demonstrated that 5-FU formation in the liver was inhibited by THU (Table 1), suggesting that 5′-DFCR is metabolized to 5′-DFUR in human liver by CDA. The genetic polymorphism of CDA has been reported, and some single nucleotide polymorphisms may affect the enzyme activity (Kirch et al., 1998; Yue et al., 2003). It has been reported that G208A (A70T), the allelic frequency of which was 4.3% in Japanese individuals, decreased 60 and 68% of the cytidine and cytarabine deaminase activities, respectively (Yue et al., 2003). Determination of the polymorphism would be important to estimate the pharmacokinetics of capecitabine.
5′-DFUR is metabolized to 5-FU by TP (Miwa et al., 1998). TP is expressed predominantly in the liver as well as in tumors (Yoshimura et al., 1990). The TP activity in colorectal tumor tissue was approximately 4-fold higher than that in the adjacent healthy tissue but was almost the same as that in the liver (Schüller et al., 2000), which is consistent with Miwa et al. (1998). In addition, the mean 5-FU concentration ratio (metastasis/healthy tissue) in the liver was reported to be 1.41 for patients with liver metastases after the administration of capecitabine (Schüller et al., 2000). Therefore, it is expected that 5′-DFUR is metabolized to 5-FU in the liver. The 5-FU formation in the liver was inhibited by TPI (Table 1), suggesting that TP is involved in the metabolism of capecitabine in the liver. Although the existence of a genetic polymorphism of TP is not known, large interindividual variability in the TP expression levels (10–320-fold) was reported in various cancer tissues, including the liver (Mori et al., 2000). The variability of the TP expression level may affect the pharmacokinetics of capecitabine.
In conclusion, we first clarified that capecitabine is metabolized to 5-FU in human liver with in vitro kinetic analyses. It was shown that CES, CDA, and TP are involved in the 5-FU formation from capecitabine in human liver. The 5′-DFCR formation in cytosol would be important as it is in microsomes. Further investigations on cytosolic enzyme will be necessary. The present study demonstrated that the bioactivation of capecitabine in the liver would be important. For the proper usage of prodrugs such as capecitabine, it will be essential to investigate the metabolic fate in the body after oral administration. This study may be helpful for the prediction of the pharmacokinetics, efficacy, and safety of capecitabine in vivo.
Acknowledgments
We thank Brent Bell for reviewing the manuscript.
Footnotes
-
This study was supported by a Grant-in-Aid for Encouragement of Young Scientists of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
-
ABBREVIATIONS: 5-FU, 5-fluorouracil; DPD, dihydropyrimidine dehydrogenase; 5′-DFUR, 5′-deoxy-5-fluorouridine (doxifluridine); CES, carboxylesterase; CDA, cytidine deaminase; TP, thymidine phosphorylase; 5′-DFCR, 5′-deoxy-5-fluorocytidine; CDHP, 5-chloro-2, 4-dihydroxypyridine; TPI, 5-chloro-6-(2-iminopyrrolidin-1-yl)methyl-2,4-(1H,3H)-pyrimidinedione; DFP, diisopropylfluorophosphate; BNPP, bis(p-nitrophenyl)phosphate; THU, tetrahydrouridine; HPLC, high-performance liquid chromatography; FBAL, α-fluoro-β-alanine.
- Received February 19, 2004.
- Accepted April 14, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}