


Abstract
Nicotine, a major constituent of tobacco, plays a critical role in smoking addiction. In humans, nicotine is primarily metabolized to cotinine, which is further metabolized to trans-3′-hydroxycotinine. Recently, we have demonstrated that heterologously expressed human CYP2A13 is highly active in the metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a nicotine-derived carcinogen. In the present study, CYP2A13-catalyzed NNK metabolism was found to be inhibited competitively by nicotine and N′-nitrosonornicotine (NNN), suggesting that both nicotine and NNN are also substrates of CYP2A13. We have further demonstrated that human CYP2A13 is indeed an efficient enzyme in catalyzing C-oxidation of nicotine to form cotinine, with the apparent Km and Vmax values of 20.2 μM and 8.7 pmol/min/pmol, respectively. CYP2A13 also catalyzes the 3′-hydroxylation of cotinine to form trans-3′-hydroxycotinine, with the apparent Km and Vmax values of 45.2 μM and 0.7 pmol/min/pmol, respectively. The importance of CYP2A13-catalyzed nicotine and cotinine metabolism in vivo remains to be determined.
Nicotine is a major constituent of tobacco, playing a critical role in establishing and maintaining tobacco dependence (Henningfield et al., 1985). The major pathway of nicotine metabolism in humans is the C-oxidation to form cotinine, which is a two-step process. The first step is catalyzed by the cytochrome P450 (P450) system to produce the intermediate nicotine-Δ-1′(5′)-iminium ion, which is further oxidized to cotinine by cytosolic aldehyde oxidase (Gorrod and Hibberd, 1982; Peterson et al., 1987; Williams et al., 1990). Other major metabolites of nicotine identified in human urine so far include trans-3′-hydroxycotinine and nicotine N′-oxide (Jacob et al., 1988). The conversion of cotinine to 3′-hydroxycotinine is highly stereospecific in vivo, with the trans isomer as a predominating product (Jacob et al., 1990). trans-3′-Hydroxycotinine is excreted in the urine to a much greater extent than cotinine itself. Cotinine may also be metabolized to 5′-hydroxycotinine, norcotinine, and cotinine N-oxide (Murphy et al., 1999).
In humans, liver is the primary site of nicotine metabolism. Nicotine is also metabolized to some extent in the lung and kidney (Benowitz et al., 1987). Previous studies demonstrated that human CYP2A6 is a key enzyme for the metabolism of nicotine to cotinine, and cotinine to trans-3′-hydroxycotinine (Flammang et al., 1992; McCracken et al., 1992; Nakajima et al., 1996a,b; Shimada et al., 1996; Messina et al., 1997). CYP2A6 is the best characterized member in the CYP2A subfamily (Pelkonen and Raunio, 1995; Fernandez-Salguero and Gonzalez, 1995; Pelkonen et al., 2000). The other two members are CYP2A7 and CYP2A13, which share a high degree of sequence identity with CYP2A6 (Fernandez-Salguero and Gonzalez, 1995). Although the transcripts of these three genes were all found in the liver, CYP2A6 was more abundant than CYP2A7 and CYP2A13 (Koskela et al., 1999). The highest level of CYP2A13 mRNA was found in human respiratory tract (Su et al., 2000). CYP2A13 mRNA was also detected in a number of human tissues, including brain, mammary gland, prostate, testis, and uterus, but not in heart, kidney, bone marrow, colon, small intestine, spleen, stomach, thymus, or skeletal muscle (Koskela et al., 1999; Su et al., 2000). We have recently demonstrated that the heterologously expressed human CYP2A13 is much more active than CYP2A6 in the metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a potent lung carcinogen (Su et al., 2000). Since NNK is derived from the nitrosation of nicotine, we predicted that nicotine would also be a substrate of CYP2A13. In the present study, we demonstrated that CYP2A13-catalyzed NNK metabolism was inhibited by nicotine in a competitive manner, and human CYP2A13 is indeed an efficient enzyme in metabolizing nicotine and cotinine.
Materials and Methods
Chemicals.trans-3′-Hydroxycotinine was kindly provided by Drs. William S. Caldwell and Gary Byrd (Targacept Inc., Winston-Salem, NC). [5-3H]NNK (purity >98%, 1.1 Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA). Unlabeled NNK and N′-nitrosonornicotine (NNN) were obtained from Chemsyn (Lenexa, KS). UV standards for NNK metabolites were a gift of Dr. Stephen Hecht (University of Minnesota, Cancer Center, Minneapolis, MN). NADPH-P450 oxidoreductase was purified from rat liver microsomes as described previously (Yasukochi and Masters, 1976). (S)-Nicotine, cotinine, NADP+, glucose 6-phosphate, and glucose-6-phosphate dehydrogenase were obtained from Sigma-Aldrich (St. Louis, MO). Tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). Other chemicals were of the highest grade available.
Heterologous Expression and Microsome Preparation. Human CYP2A13 cDNA was subcloned into a baculovirus expression vector and expressed in Sf9 insect cells (Su et al., 2000). Microsomes were prepared by differential centrifugation, and the microsomal protein content was determined using a Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA). Total P450 content was determined as described previously (Omura and Sato, 1964).
Inhibition of NNK Metabolism. Incubation conditions for NNK metabolism were the same as those described previously (Su et al., 2000) except for the addition of the inhibitors, nicotine and NNN. Briefly, the incubation mixture contained 100 mM sodium phosphate buffer (pH 7.4), 5 mM sodium bisulfite, 1 mM EDTA, 3 mM magnesium chloride, an NADPH-generating system (1 mM NADP+, 5 mM glucose 6-phosphate, 1.5 units of glucose-6-phosphate dehydrogenase), NADPH-P450 oxidoreductase, 10 pmol of CYP2A13 (reductase/P450 = 5:1 in molar ratio), 10 μM NNK (containing 1 μCi of [5-3H]NNK), and 40 μM nicotine or 30 μM NNN in a total volume of 0.4 ml. The nicotine and NNN concentrations selected for the inhibition study were based on the known Km values of CYP2A13 for NNK metabolism (∼10 μM for both keto aldehyde and keto alcohol formation; Su et al., 2000). After a 15-min incubation, the reaction was terminated by addition of zinc sulfate and barium hydroxide. The sample was centrifuged and filtered, and an aliquot (0.2 ml) was injected onto a high-performance liquid chromatograph for the analysis of NNK metabolites.
Nicotine and Cotinine Metabolism. The metabolism of nicotine and cotinine as well as the subsequent analysis of their metabolites was conducted as described (Nakajima et al., 1996a,b), with slight modifications. The incubation mixture (0.2 ml final volume) consisted of 50 mM Tris-HCl buffer (pH 7.4), 5 mM magnesium chloride, 5 mM EDTA, the NADPH-generating system, rat liver cytosol (as a source of cytosolic aldehyde oxidase for cotinine formation), NADPH-P450 oxidoreductase, 30 to 50 pmol of expressed CYP2A13 (same reductase/P450 ratio as above), and nicotine or cotinine. The reaction was initiated by the addition of NADPH-generating system after a 2-min preincubation at 37°C and was terminated at the end of a 30-min incubation by the addition of ice-cold acetone. After vortex mixing and centrifugation to remove the protein, the resulting supernatant was extracted three times (4 ml total volume) with 2-propanol/methylene chloride (1:2, v/v). The organic fraction was separated by centrifugation and evaporated under a stream of nitrogen at 40°C. The residue was reconstituted in 100 μl of the mobile phase, and 80 μl of it was injected onto the high-performance liquid chromatograph. For quantification of the metabolites, a known amount of cotinine or trans-3′-hydroxycotinine was used as internal standard and was added to the incubation mixture after the enzyme was inactivated by acetone, followed by extraction and analyses similar to those of the samples. The recovery of cotinine and 3-hydroxycotinine was approximately 78%. Microsomes prepared from the Sf9 cells infected with the expression vector only (without CYP2A13 cDNA) were used as a negative control. For enzyme kinetic studies, at least seven different substrate concentrations were used in the incubations.
HPLC Analysis. HPLC analysis was performed with a Waters system (Waters, Milford, MA) consisting of model 501 pumps, a model 710B autosampler, and a model 712 integrator. The system was coupled to a Radiomatic Flo-One/Beta radioflow detector (Radiomatic Instruments & Chemical Company, Tampa, FL) for the analysis of NNK metabolites, and to a 490E Waters PDA detector for detecting the nicotine and cotinine metabolites. The HPLC conditions for the analysis of NNK metabolites were the same as those described previously (Hong et al., 1992). A μBondapak C18 column (3.9 × 300 mm) was used for analysis of the nicotine and cotinine metabolites. Mobile phase A contained 10 mM potassium phosphate (pH 7.0) and 0.1% triethylamine; mobile phase B consisted of acetonitrile and 2-propanol (7:3, v/v). The column was eluted isocratically using 88% A and 12% B for nicotine metabolite analysis, and with 98.5% A and 1.5% B for cotinine metabolite analysis. The eluent was monitored at A260. The flow rate was maintained at 0.8 ml/min. Under these conditions, cotinine was eluted at 5 min, and trans-3′-hydroxycotinine eluted at 11 min.
Data Analysis. Kinetic parameters (Km and Vmax) were determined by curve fitting and nonlinear regression analysis using the Enzyme Kinetics V1.11 software (Trinity Software, Plymouth, NH). The values are mean ± S.D. (n = 3) or the average of two separate experiments.
Results and Discussion
Consistent with our previous study, the heterologously expressed human CYP2A13 is highly efficient in catalyzing the α-hydroxylation of NNK, leading to the formation of keto aldehyde (from α-hydroxylation of the methylene carbon) and keto alcohol (from α-hydroxylation of the methyl carbon). As shown in Table 1, our previously reported Km and Vmax values of CYP2A13 (Su et al., 2000) were confirmed by the present study. When nicotine or NNN, another nicotine-derived carcinogen, was added to the incubation mixture, the CYP2A13-catalyzed formation of keto alcohol and keto aldehyde was significantly inhibited. Figure 1 shows the effects of nicotine and NNN on the kinetics of keto aldehyde formation by CYP2A13. The inhibition is competitive in nature, since the addition of nicotine or NNN caused a 4- to 6-fold increase in the Km value but did not affect the Vmax (Table 1).
Effects of nicotine and NNN on the kinetic parameters of CYP2A13-catalyzed NNK metabolism
Values in the parentheses were obtained from our previous report (Su et al., 2000), which agree well with the present result.
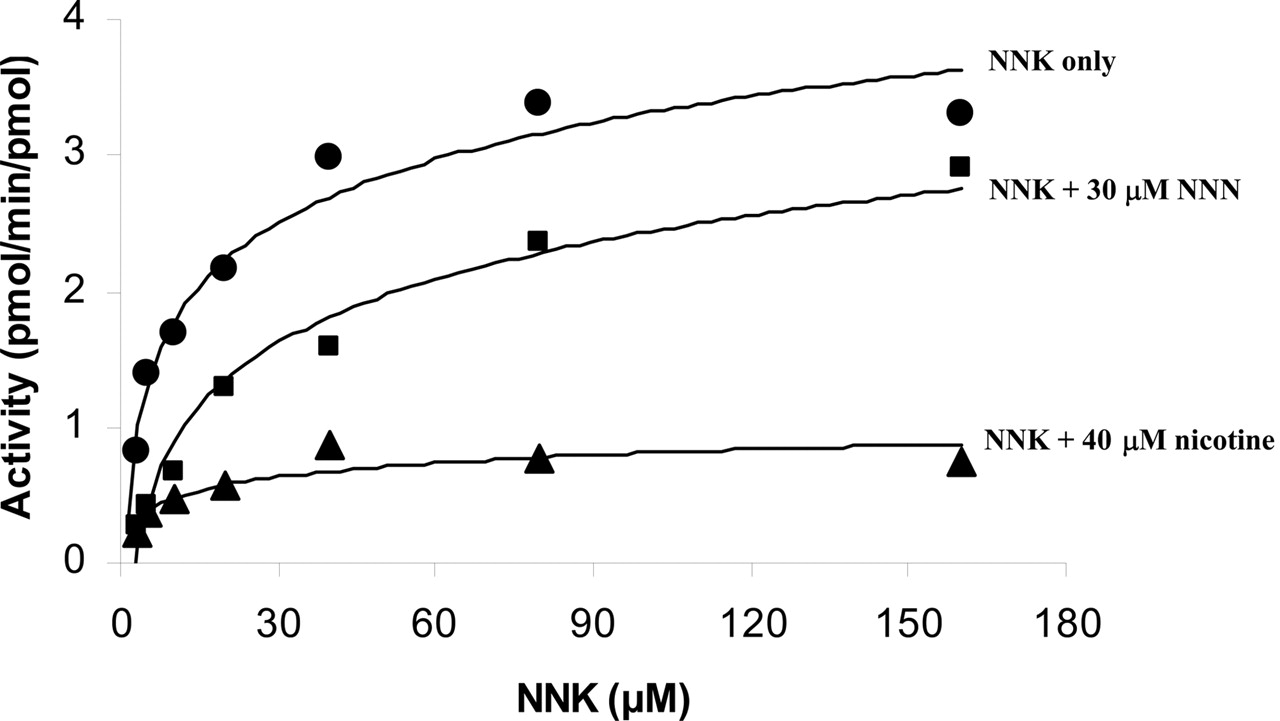
Kinetics of CYP2A13-catalyzed NNK metabolism (keto aldehyde formation) and the effect of nicotine and NNN. NNK (10 μM) was incubated with 10 pmol of CYP2A13 either alone or in the presence of nicotine (40 μM) or NNN (30 μM) at 37°C for 15 min.
The competitive inhibition of NNK metabolism by nicotine and NNN is a strong indication that both nicotine and NNN are also substrates of CYP2A13. We therefore directly determined the activity of CYP2A13 in metabolizing nicotine and cotinine. After optimization of the assay conditions, the kinetics of cotinine and trans-3′-hydroxycotinine formation by CYP2A13 were determined at different substrate concentrations, ranging from 5 to 300 μM nicotine (for nicotine metabolism) and 25 to 1000 μM cotinine (for cotinine metabolism) (Fig. 2). As shown in Table 2, CYP2A13 is efficient in catalyzing nicotine metabolism, with the Km value of 20.2 μM and Vmax of 8.7 pmol/min/pmol P450. CYP2A13 is also active in catalyzing cotinine metabolism, with the Km value of 45.2 μM and Vmax of 0.67 pmol/min/pmol P450. Table 2 also compared CYP2A13 with CYP2A6 for the catalytic efficiency in the metabolism of nicotine and cotinine. Corresponding kinetic parameters of CYP2A6 were obtained from reported studies in which the metabolism of nicotine and cotinine by lymphoblastoid cell-expressed CYP2A6 was assayed under similar conditions (Nakajima et al., 1996a,b). The Vmax values for CYP2A6 were converted from the original data, based on the reported P450 content in the microsomal proteins. The catalytic efficiency (Vmax/Km) of CYP2A13 is apparently much higher than that of CYP2A6 for both nicotine and cotinine metabolism. Although the Km value for nicotine metabolism is not significantly different between CYP2A13 and CYP2A6 (20.2 μM versus 26 μM), CYP2A13 has a much lower Km value than CYP2A6 for cotinine metabolism (45.2 μM versus 265 μM).

Kinetics of CYP2A13-catalyzed metabolism of nicotine (A) and cotinine (B). Nicotine or cotinine at different concentrations was incubated with 30 to 50 pmol of CYP2A13 at 37°C for 30 min.
Kinetic parameters of nicotine and cotinine metabolism by human CYP2A13 and CYP2A6
For nicotine metabolism, the Km and Vmax values of CYP2A13 are the average of two separate experiments. Values of each experiment are shown in parentheses. For cotinine metabolism, the Km and Vmax values of CYP2A13 are the mean ± S.D. of three separate experiments.
In contrast to CYP2A6, which is mainly expressed in the liver, CYP2A13 is predominantly expressed in human respiratory tract. CYP2A13 is also expressed in many other nonhepatic human tissues including brain (Su et al., 2000). Since liver is the primary site for the metabolism of xenobiotics including nicotine, the low level of CYP2A13 expression in the human liver suggests that CYP2A13 plays an insignificant role in the metabolism-related clearance of nicotine in the human body, even though its catalytic efficiency is higher than that of CYP2A6 in nicotine metabolism. This notion is supported by a recent observation on the metabolism of nicotine in individuals who were homozygous for CYP2A6 gene deletion. After the same dose exposure of nicotine, their cumulated urinary cotinine level was only approximately one-seventh of that in the control subjects who carried wildtype CYP2A6 gene (Kitagawa et al., 1999). This clearly indicates that CYP2A6 is the major enzyme responsible for nicotine clearance in vivo. Similarly, the CYP2A13-catalyzed cotinine metabolism would not be expected to contribute significantly to the pharmacokinetic profile of cotinine in the human body. However, although CYP2A13 may not be involved in the clearance of nicotine and cotinine, it is tempting to speculate that CYP2A13-catalyzed nicotine and cotinine metabolism in the nonhepatic target tissues of nicotine may play an important role in nicotine-induced toxic responses. The concentration threshold of nicotine to induce toxic responses in the target tissues and/or cells might be quite different, and an efficient metabolism in situ by CYP2A13 may affect the threshold.
Due to its high efficiency in the metabolic activation of tobacco-specific carcinogen NNK and its predominant expression in human respiratory tract, CYP2A13 is believed to play a significant role in smoking-related human respiratory cancers (Su et al., 2000; Wang et al., 2003). Although nicotine has not been directly linked to the initiation of lung cancer, up-regulation of the nicotinic receptor pathway by nicotine and nicotine-derived NNK and NNN has been proposed to be an important factor in the development of small cell lung carcinoma in smokers (Plummer et al., 2000; Schuller et al., 2000). Nicotine at low concentration (nanomolar) was also found to activate the mitogen-activated protein kinase and the protein kinase C pathways in lung cancer cell lines, leading to the inhibition of apoptosis (Heusch and Maneckjee, 1998). It would be of interest to study the effect of CYP2A13-mediated nicotine metabolism in situ on the development of small cell lung carcinoma and on other nicotine-induced responses in the nicotine target tissues such as brain.
Acknowledgments
We thank Drs. William S. Caldwell and Gary Byrd (Targacept Inc., Winston-Salem, NC) for the gift of trans-3′-hydroxycotinine standard, Mao-Jung Lee (Rutgers University, Piscataway, NJ) for advice on HPLC analysis, and Wen-Yu Hu for helpful suggestions.
Footnotes
-
Part of this work was supported by National Institutes of Health Grant R01-ES10048 (J.-Y.H.), R01-CA 092596 (X.D.) and National Institute of Environmental Health Sciences Center Grant ES05022.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002105.
-
ABBREVIATIONS: P450, cytochrome P450; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; NNN, N′-nitrosonornicotine; HPLC, high-performance liquid chromatography.
- Received August 30, 2004.
- Accepted November 3, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}