


Abstract
Breast cancer resistance protein (BCRP) is a recently identified ATP-binding cassette transporter, important in drug disposition and in the development of multidrug resistance in cancer. Flavonoids, a major class of natural compounds widely present in foods and herbal products, have been shown to be human BCRP inhibitors. The objective of the present study was to evaluate the potential for in vivo pharmacokinetic interactions by comparing the pharmacokinetics of topotecan (a model BCRP substrate) after oral administration of 2 mg/kg topotecan with and without different doses of the flavonoids chrysin or 7,8-benzoflavone (BF) in rats and mdr1a/1b (–/–) mice. Coadministration of 50 mg/kg GF120918 [N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9, 10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide] with 2 mg/kg topotecan significantly increased the area under the plasma concentration-time curve and bioavailability of topotecan by more than 4-fold in these animals, indicating the importance of BCRP in the bioavailability and disposition of topotecan in rats. Chrysin (50 μM) and BF (5 μM) significantly inhibited the BCRP-mediated transport of topotecan in BCRP-overexpressing MCF-7 MX100 cells (MCF-7 cells selected with mitoxantrone) to a level comparable to that observed with 10 μM fumitremorgin C (a potent BCRP inhibitor). However, neither chrysin nor BF significantly altered topotecan pharmacokinetics in rats or in mdr1a/1b (–/–) mice after oral coadministration of doses up to 50 mg/kg. The reason(s) for this lack of in vitro-in vivo association may be the lack of potent inhibition activity of the flavonoids against mouse or rat BCRP, as evidenced by our observation that these flavonoids have only weak, if any, inhibition activity against mouse Bcrp1-mediated transport of topotecan in MDCK-Bcrp1 cells.
Flavonoids are a large class of polyphenolic compounds present in vegetables, fruits, and plant-derived beverages, and in many herbal preparations marketed as over-the-counter dietary supplements. The average intake of total flavonoids from the Western diet was estimated to be 1 g per day (Kuhnau, 1976), although this may be an overestimation and the actual intake may be much lower. Numerous studies have indicated that flavonoids have antioxidant, anticarcinogenic, antiviral, anti-inflammatory, and antiestrogenic (estrogenic) activities (Middleton et al., 2000; Havsteen, 2002), suggesting that flavonoids may play a protective role in the prevention of cancer, coronary heart diseases, bone loss, and many other age-related degenerative diseases (Hertog et al., 1993; Havsteen, 2002). As such, the consumption of flavonoid-containing products is becoming more and more wide-spread with the burgeoning public interest in alternative medicine and in disease prevention. However, significant or even life-threatening pharmacokinetic interactions of flavonoids or flavonoid-containing food/herbal products with conventional drugs have been observed in animal and clinical studies (Ruschitzka et al., 2000; Dresser et al., 2002). For example, in vitro studies have reported that flavonoids are P-glycoprotein and CYP3A inhibitors (Conseil et al., 1998; Ho et al., 2001), and the flavonoids flavone and quercetin have been shown to increase the bioavailability of paclitaxel (P-glycoprotein and CYP3A substrate) in rats in a flavonoid dose-dependent manner (Choi et al., 2004a,b). Interestingly, this in vitro-in vivo association was not observed when the flavonoids quercetin and phellamurin were coadministered with cyclosporin A, which is also a substrate of both P-glycoprotein and CYP3A; in fact, the bioavailability of cyclosporin A was significantly decreased by the coadministration of these flavonoids in pigs and/or rats (Chen et al., 2002; Hsiu et al., 2002). The reason(s) for this apparent in vitro-in vivo inconsistency is still unknown. The potential for flavonoid-drug interactions and the widespread use of flavonoid-containing herbal products emphasizes the need for further in vivo studies to evaluate clinically relevant pharmacokinetic drug interactions.
Recently, we and other investigators have demonstrated in cell culture studies that many flavonoids can inhibit human breast cancer resistance protein (BCRP, ABCG2) (Cooray et al., 2004; Imai et al., 2004; Zhang et al., 2004b). BCRP is a recently identified plasma membrane efflux transporter belonging to the ATP-binding cassette transporter superfamily (Litman et al., 2001). The mouse ortholog of BCRP (Bcrp1) has also been cloned and shown to share 81% amino acid sequence identity with human BCRP (Allen et al., 1999). Many important drugs, such as topoisomerase I inhibitors, mitoxantrone, methotrexate, flavopiridol, and nucleoside human immunodeficiency virus reverse transcriptase inhibitors, have been shown to be BCRP substrates (Doyle and Ross, 2003). Apart from its expression in a number of human tumors (Doyle and Ross, 2003), which suggested its potential role in multidrug resistance (Steinbach et al., 2002), BCRP is also expressed in the excretory organs and tissues with barrier functions, such as liver canalicular membranes, the luminal surface of intestine, blood-brain barrier, and human placenta (Maliepaard et al., 2001; Cooray et al., 2002), and has been shown to play an important role in drug disposition. For example, coadministration of GF120918 (a BCRP inhibitor) significantly increased the bioavailability and decreased the biliary excretion of topotecan (a BCRP substrate) in mdr1a/1b (–/–) mice and in humans (Jonker et al., 2000; Kruijtzer et al., 2002). Therefore, inhibition of BCRP by flavonoids is hypothesized to increase the bioavailability of drugs that are BCRP substrates.
The objective of the present study was to test the above hypothesis by investigating the effects of the flavonoids chrysin and 7,8-benzoflavone (BF) (Fig. 1) on topotecan pharmacokinetics in rats and in mdr1a/1b (–/–) mice. Topotecan has been shown to be a very good BCRP substrate but a poor (or moderate) P-glycoprotein substrate (Jonker et al., 2000), and its bioavailability has been shown to be dramatically changed by inhibition of BCRP (Jonker et al., 2000; Kruijtzer et al., 2002). In addition, topotecan only undergoes very limited metabolism in rats and humans (Rosing et al., 1997; Platzer et al., 1998), and the plasma peak concentration of its major metabolite, N-desmethyl topotecan, was only 0.7% of the peak concentration of total topotecan (both the lactone form and the carboxylate form) after a 30-min infusion of topotecan to patients (Rosing et al., 1997). Therefore, using topotecan as a model BCRP substrate drug can simplify the interpretation by avoiding the confounding factors that could result from the interaction of flavonoids with drug-metabolizing enzymes. The flavonoids chrysin and BF are both potent BCRP inhibitors, and their EC50 values for inhibiting BCRP in MCF-7 cells selected with mitoxantrone (MCF-7 MX100 cells) (measured as the concentration required to produce 50% of the maximal increase of mitoxantrone accumulation in MCF-7 MX100 cells, which overexpress wild-type BCRP) were shown to be 0.39 ± 0.13 μM (Zhang et al., 2004a) and 0.07 ± 0.02 μM (unpublished data from this laboratory), respectively.

The chemical structures of chrysin and 7,8-benzoflavone (BF).
Materials and Methods
Materials. Chrysin, acridine, glycofurol, phosphoric acid, and triethylamine were purchased from Sigma-Aldrich (St. Louis, MO); and BF was purchased from Indofine (Hillsborough, NJ). Topotecan was purchased from ChemPacific (Baltimore, MD). RPMI 1640, Dulbecco's modified Eagle's medium, fetal bovine serum, phosphate-buffered saline (PBS), and Hanks' buffered salt solution (HBSS) were purchased from Invitrogen (Carlsbad, CA). [3H]Mannitol (15 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). GF120918 and PSC833 were kind gifts from GlaxoSmithKline (Research Triangle Park, NC) and Novartis (Basel, Switzerland), respectively. Human breast cancer MCF-7/sensitive and MCF-7 MX100 cells, as well as fumitremorgin C (FTC), were kind gifts from Dr. Susan E. Bates (National Cancer Institute, Bethesda, MD). MDCK-mock (MDCK cells transfected with empty vector) and MDCK-Bcrp1 (MDCK cells transfected with mouse Bcrp1) cells were kind gifts from Dr. Alfred Schinkel (Netherlands Cancer Institute, Amsterdam, The Netherlands). All the other reagents or solvents used were either analytical or high-performance liquid chromatography (HPLC) grade.
Cell Culture. MCF-7 cells (both sensitive and resistant) were cultured in 75-cm2 flasks with RPMI 1640 culture medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2/95% air. The culture medium also contained 100 units/ml penicillin and 100 μg/ml streptomycin. For MCF-7 MX100 cells, the culture medium also contained 100 nM mitoxantrone. MCF-7 MX100 cells have been shown to overexpress human wild-type BCRP with no detectable expression of P-glycoprotein or MRP1 (Robey et al., 2004; Zhang et al., 2004b); MCF-7/sensitive cells have no significant expression of any of these transporters (Zhang et al., 2004b). MDCK-mock and MDCK-Bcrp1 cells were cultured in 75-cm2 flasks with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2/95% air. The culture medium also contained 100 units/ml penicillin and 100 μg/ml streptomycin.
Topotecan Accumulation Studies. The accumulation studies were performed after the cells reached 90% confluence in 35-mm2 dishes. Cells were washed twice with 1× PBS and then incubated with 1 ml of RPMI 1640, containing 5 μM topotecan and specified concentrations of the test compounds, or 0.1% DMSO (as control). FTC is a specific BCRP inhibitor (Rabindran et al., 1998) and was used as the positive control in these experiments. After a 10-min incubation, topotecan accumulation was stopped by rinsing the cells four times with ice-cold PBS. Cells were then harvested into 200 μl of distilled water and lysed by sonication. Aliquots of the cell lysates were used to determine topotecan concentration by HPLC and protein content using the BCA protein assay. Topotecan accumulation was normalized for the protein content and was expressed as percentage accumulation in the control group.
Animals. Female Sprague-Dawley (SD) rats (body weight 180–220 g) were purchased from Harlan (Indianapolis, IN) and male mdr1a/1b (–/–) mice (body weight 23.0–26.5 g) were purchased from Taconic Farms (Germantown, NY). The animals were housed in a 12-h light/12-h dark environment with free access to tap water and standard chow. Animals were acclimatized to this environment for at least 1 week, and rats had right jugular vein cannulation following an intramuscular injection of 90 mg/kg ketamine (Henry Schein, Melville, NY) and 10 mg/kg xylazine (Henry Schein) at least 3 days before the study. The study was approved by the University at Buffalo Institutional Animal Care and Use Committee.
Pharmacokinetic Studies.Rat Studies. The SD rats were fasted overnight and, then, a specified dose of the test compound (GF120918, chrysin, or BF dissolved in glycofurol) or the vehicle glycofurol (as control) was administered orally (by gavage). The test compounds were dissolved in glycofurol at such a concentration that the specified dose was delivered as 2.5 μl/g body weight drug solution. Three minutes later, the animals were administered 2 mg/kg topotecan orally (0.8 mg/ml dissolved in saline containing 5% d-glucose). For the calculation of topotecan bioavailability, three rats without prior drug administration were given an i.v. dose of 1 mg/kg topotecan (0.8 mg/ml dissolved in saline containing 5% d-glucose). The blood samples (150 μl) were taken from the jugular vein cannula at 0 (before topotecan administration), 2, 7, 15, 30, 60, 120, 240, 480, and 720 min after topotecan administration. The plasma samples were obtained by centrifugation and stored at –80°C until drug analysis. During the experiment, the rats had free access to water at all times and free access to food (standard chow) beginning 4 h after topotecan administration.
mdr1a/1b (–/–) Mouse Studies. The mice were fasted overnight and then administered 50 mg/kg chrysin (suspended in olive oil at a concentration of 5 mg/ml) by administering 10 μl/g body weight chrysin suspension or the vehicle olive oil (as control) orally (by gavage). Three minutes later, the mice were given 2 mg/kg topotecan (0.2 mg/ml dissolved in saline containing 5% d-glucose) orally. The blood samples (20 μl) were taken from the retro-orbital plexus at 0 (before topotecan administration), 15, 30, 60, 180, and 360 min after topotecan administration. The mouse plasma was obtained by centrifugation and stored at –80°C until analysis. During the experiment, the mice had free access to water and, 4 h after topotecan administration, free access to mouse chow.
Topotecan Bidirectional Transport Studies. Transport experiments using MDCK-Bcrp1 cell monolayers were performed in a manner similar to that previously described (Zhang and Morris, 2003). Briefly, MDCK-bcrp1 cells were seeded in Transwell polycarbonate inserts (six-well, 0.4-μm pore size; Corning Glassworks, Corning, NY) at a density of approximately 106 cells/well and grown for 7 days. During the study, the cell monolayers were first washed with HBSS (pH 7.2) twice for a total of 30 min. Transport buffer (HBSS, pH 7.2) containing specified concentrations of the test compounds or 0.1% DMSO (control) was then loaded in both apical (1.5 ml) and basolateral (2.6 ml) chambers and incubated at 37°C. After 30 min, topotecan (10 μM) was added to either the apical or basolateral chamber (donor chamber), and the incubation was continued. The samples (150 μl) were taken from the opposite chamber (receiver chamber) at 30, 60, and 90 min after addition of topotecan and replaced with the same volume of fresh transport buffer. To ensure the monolayer integrity, the apparent permeability coefficients (Papp) of [3H]mannitol, a paracellular marker, across MDCK-Bcrp1 cell monolayers in both apical to basolateral (AP-to-BL) and basolateral to apical (BL-to-AP) directions were also measured in triplicate, in parallel, and these values were all lower than 1.00 × 10–6 cm/s.
Topotecan Analysis.Analysis of Topotecan in Rat Plasma, Cell Lysates, and Buffer Samples. The plasma concentration of total topotecan (both the lactone form and carboxylate form) in rat plasma, cell lysates from topotecan accumulation studies and samples from topotecan bidirectional transport studies was assayed by a validated HPLC method with the lower limit of quantitation of 0.02 ng on column (Rosing et al., 1999; Chen and Balthasar, 2002) with minor modifications. Briefly, an aliquot of 40 μl sample was spiked with 4 μl of 1.5 μg/ml acridine (in methanol), and then 120 μl methanol and 40 μl of 100 mM phosphoric acid were added and mixed. The mixture was then centrifuged and 50 μl of the supernatant was injected for HPLC analysis. The HPLC analysis was carried on a Partisphere C18 column (partisphere 5 μm C18, 125 × 4.6 mm i.d., Phenomenex) with a flow rate of 1 ml/min. The mobile phase was 10 mM phosphate buffer pH 3.74, 25% methanol, 2% trimethylamine. Topotecan was detected with a Waters 474 Scanning Fluorescence detector at an excitation wavelength of 361 nm and emission wavelength of 527 nm. For rat plasma samples, this HPLC assay has an intraday variability less than 8.57% and an interday variability less than 13.7%. Standard curves are linear over the concentration range of 1–500 ng/ml.
Analysis of Topotecan in Mouse Plasma. Topotecan concentrations in mouse plasma were determined after the addition of 200 μl of 50:50 methanol/acetonitrile to 5 μl of plasma sample and 10 μl of acridine (internal standard, 500 ng/ml) to precipitate proteins. The mixture was then centrifuged, and the supernatant was removed and dried under nitrogen at 50°C. The sample was then resuspended in 35 μl of 25 mM phosphoric acid and 35 μl of 50;50 methanol/acetonitrile. The sample was injected onto a Zorbax SB-C18 column (75 × 4.6 mm i.d., particle size 3.5 μm) protected with a stainless steel frit (2.0 μm). The mobile phase was pumped through the column with a flow rate of 0.65 ml/min, and consisted of acetonitrile and 10 mM ammonium formate containing 0.1% formic acid. The ions were measured using an API3000 spectrometer (PerkinElmerSciex Instruments, Boston, MA) with conditions that were optimized for both topotecan and acridine. The lower limit of quantitation of topotecan was 1 ng/ml.
Pharmacokinetic Analysis. The pharmacokinetic parameters of topotecan were obtained by noncompartmental analysis using WinNonlin version 2.1 (Pharsight, Mountain View, CA). The area under the plasma concentration-time curve (AUC) was calculated using the trapezoidal method; the terminal half-life (t1/2) of topotecan was calculated as 0.693/k, and k was determined from the slope of the terminal regression line. The bioavailability (F) of topotecan after oral administration was calculated by the following equation:  where AUC0-∞ is the AUC from time 0 to infinity.
where AUC0-∞ is the AUC from time 0 to infinity.
Calculation of the Papp of Topotecan across MDCK Cells. The apparent permeability coefficients (Papp) across MDCK-mock and MDCK-Bcrp1 cell monolayers in both the AP-to-BL (Papp,AP-BL) and BL-to-AP (Papp,BL-AP) directions were calculated as follows:  where ΔQ/Δt is the rate of topotecan appearing in the receiver chamber, which was obtained as the slope of the regression line on the transport-time profile of topotecan across the cell monolayers, C0 is the initial concentration of topotecan loaded in the donor chamber, and A is the cell monolayer surface area (4.71 cm2).
where ΔQ/Δt is the rate of topotecan appearing in the receiver chamber, which was obtained as the slope of the regression line on the transport-time profile of topotecan across the cell monolayers, C0 is the initial concentration of topotecan loaded in the donor chamber, and A is the cell monolayer surface area (4.71 cm2).
Statistical Analysis. The differences between the mean values were analyzed for significance using Student's t test or one-way analysis of variance, followed by Dunnett's test. p values <0.05 were considered statistically significant.
Results
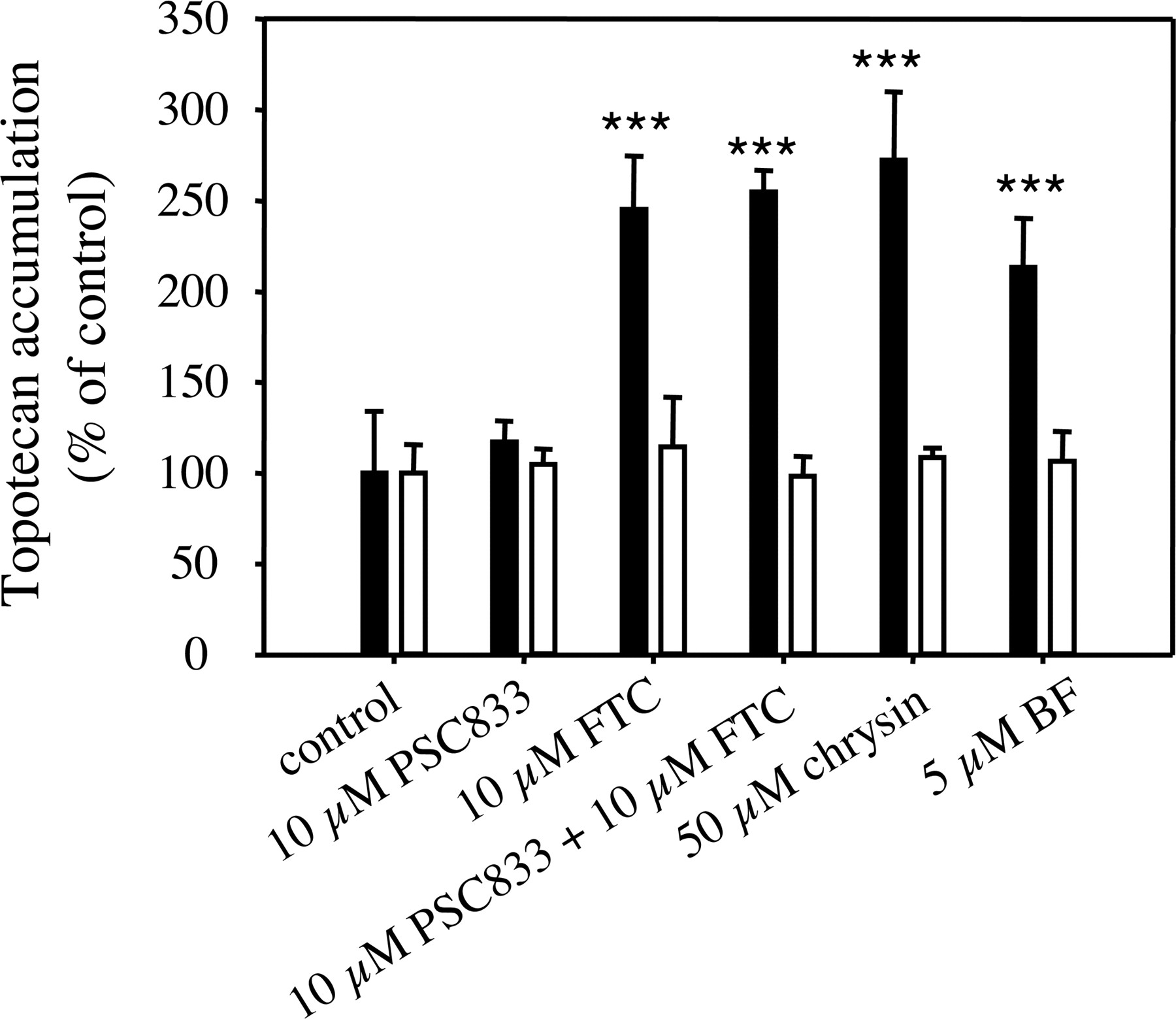
Topotecan Accumulation in MCF-7/Sensitive and MCF-7 MX100 Cells. The flavonoids chrysin (Zhang et al., 2004b) and BF have been shown to inhibit BCRP-mediated efflux of mitoxantrone; however, their effects on BCRP-mediated transport of topotecan have not been evaluated. Therefore, we conducted a study to investigate the effects of chrysin and BF on topotecan accumulation in both BCRP-negative MCF-7/sensitive cells and BCRP-overexpressing MCF-7 MX100 cells. As we can see from Fig. 2, none of the tested compounds (FTC, PSC833, chrysin, and BF) at the specified concentrations had significant effects on topotecan accumulation in MCF-7/sensitive cells. However, in the resistant MCF-7 MX100 cells, the BCRP inhibitor FTC (10 μM), when used alone or in combination with 10 μM PSC833, which is a P-glycoprotein inhibitor with negligible effect on BCRP-mediated transport (Jonker et al., 2000), significantly increased topotecan accumulation to a similar level [245 ± 29.7%, p < 0.001 (alone) and 255 ± 11.8%, p < 0.001 (in combination), respectively]. PSC833 (10 μM) administered alone had no significant effect on topotecan accumulation (105 ± 8.51%, p > 0.05), indicating that the topotecan accumulation in MCF-7 MX100 cells was limited by the expression of BCRP in these cells. Chrysin (50 μM) and BF (5 μM) both significantly increased topotecan accumulation in MCF-7 MX100 cells (272 ± 37.5%, p < 0.001 and 213 ± 27.0%, p < 0.001, respectively), and the average increases of topotecan accumulation produced by 50 μM chrysin and 5 μM BF reached 119% and 78% of that produced by 10 μM FTC, indicating that these flavonoids can potently inhibit the BCRP-mediated efflux of topotecan.

Effects of flavonoids chrysin and BF on topotecan accumulation in human breast cancer MCF-7 cells. The 10-min accumulation of topotecan (5 μM) in MCF-7/sensitive (open bars) and MCF-7 MX100 cells (black bars) in the presence of 0.1% DMSO (control) or the test compounds at specified concentrations was determined as described under Materials and Methods. The accumulation of topotecan was expressed as percentage of the control accumulation. Data are expressed as mean ± S.D., n = 4. ★★★, p < 0.001.
Effects of GF120918 on Topotecan Pharmacokinetics in SD Rats. The oral bioavailability and biliary clearance of topotecan in mice and humans are believed to be affected by the expression of BCRP because coadministration of GF120918 (a BCRP inhibitor) has been shown to increase the AUC of topotecan by more than 6-fold in mdr1a/1b (–/–) mice (Jonker et al., 2000) and to increase the apparent bioavailability of topotecan from 40% to 97% in humans (Kruijtzer et al., 2002) after oral administration. To confirm that the bioavailability of topotecan in rats is also limited by the expression of BCRP, we conducted a study to evaluate the effects of GF120918 on topotecan pharmacokinetics in SD rats. As can be observed from Fig. 3 and Table 1, coadministration of 50 mg/kg GF120918 significantly increased the AUC0–720 (the AUC from time 0 to 720 min, which is the last time point for blood sampling) [from 1.74 ± 0.86 (×104) to 7.65 ± 3.78 (×104) ng/ml · min, p < 0.01], the AUC0-∞ [from 1.80 ± 0.89 (×104) to 7.91 ± 3.56 (×104) ng/ml · min, p < 0.01], and the bioavailability (from 29.7 ± 14.8% to 130 ± 58.8%, p < 0.01) by more than 4-fold. The mean Cmax was also increased 3-fold (from 86.4 ± 42.9 to 257 ± 154 ng/ml, p < 0.05) (Fig. 3; Table 1). The tmax and terminal t1/2 were not significantly changed by GF120918 (Fig. 3; Table 1). These results indicate that the bioavailability of topotecan in rats is indeed limited by the expression of BCRP and, therefore, inhibition of BCRP should increase the bioavailability of topotecan in these animals.

Plasma concentration-time profile of topotecan after the administration of topotecan (2 mg/kg) alone or with GF120918 (50 mg/kg) in SD rats. The plasma concentration-time profile of topotecan after an oral dose of 2 mg/kg topotecan in SD rats (control, •) or with 50 mg/kg GF120918 (▪), administered 3 min before topotecan, was determined as described under Materials and Methods. For the calculation of bioavailability, the plasma concentration-time profile of topotecan in SD rats after i.v. bolus injection of 1 mg/kg topotecan (♦) was also determined. Data are expressed as mean ± S.E.; for control, n = 7, for GF120918, n = 4, and for the i.v. dose of topotecan, n = 3.
Pharmacokinetic parameters of topotecan in SD rats after oral administration of topotecan (2 mg/kg) either alone or with GF120918, chrysin, and BF
The plasma concentration-time profile of topotecan in SD rats after oral coadministration of topotecan (2 mg/kg) with glycofurol (control) or the specified doses of the test compounds was determined as described under Materials and Methods. The pharmacokinetic parameters were obtained by noncompartmental analysis using WinNonlin. Data are expressed as mean ± S.D. The number of animals for various treatment groups is specified in parentheses after the corresponding group names.
Effects of Chrysin on Topotecan Pharmacokinetics in SD Rats. Chrysin has been shown to be a potent BCRP inhibitor, and it significantly inhibited the BCRP-mediated transport of topotecan in MCF-7 MX100 cells at a 50 μM concentration; therefore, the coadministration of chrysin with topotecan would be expected to increase topotecan oral bioavailability. However, as we can see from Table 1, none of the pharmacokinetic parameters of topotecan in the SD rats were significantly changed by the coadministration of either 5 mg/kg or 50 mg/kg chrysin with an oral dose of 2 mg/kg topotecan. Actually, instead of increasing the AUC and bioavailability of topotecan, chrysin decreased the AUC0–720 of topotecan in a chrysin dose-dependent manner, although these decreases were not statistically significant.
Effects of Chrysin on Topotecan Pharmacokinetics in mdr1a/1b (–/–) Mice. To confirm the observations in the rat study, we also looked at the effects of chrysin on topotecan pharmacokinetics in mdr1a/1b (–/–) mice after oral coadministration of 50 mg/kg chrysin with 2 mg/kg topotecan. As can be seen from Table 2, consistent with the rat study findings, none of the pharmacokinetic parameters of topotecan were significantly changed by chrysin administration in mdr1a/1b (–/–) mice.
Pharmacokinetic parameters of topotecan in mdr1a/1b (-/-) mice after oral coadministration of topotecan (2 mg/kg) with the vehicle olive oil (control) or chrysin (50 mg/kg)
The plasma concentration-time profile of topotecan in mdr1a/1b (-/-) mice after oral coadministration of topotecan (2 mg/kg) with olive oil (control) or 50 mg/kg chrysin was determined as described under Materials and Methods. The pharmacokinetic parameters were obtained by noncompartmental analysis using WinNonlin. Data are expressed as mean ± S.D. n = 4 for both control and chrysin groups.
Effects of BF on Topotecan Pharmacokinetics in SD Rats. In addition to chrysin, we also determined the effects of BF on topotecan pharmacokinetics in SD rats. This compound has higher inhibition activity than chrysin on BCRP-mediated transport (EC50 value, 0.07 ± 0.02 μM), and it significantly increased topotecan accumulation in MCF-7 MX100 cells at a 5 μM concentration. As shown in Table 1, although the AUC0–720, AUC0-∞,Cmax, and bioavailability of topotecan were slightly increased by the coadministration of BF in a dose-dependent manner, none of these pharmacokinetic parameters were significantly changed by the coadministration of either 10 mg/kg or 50 mg/kg BF.
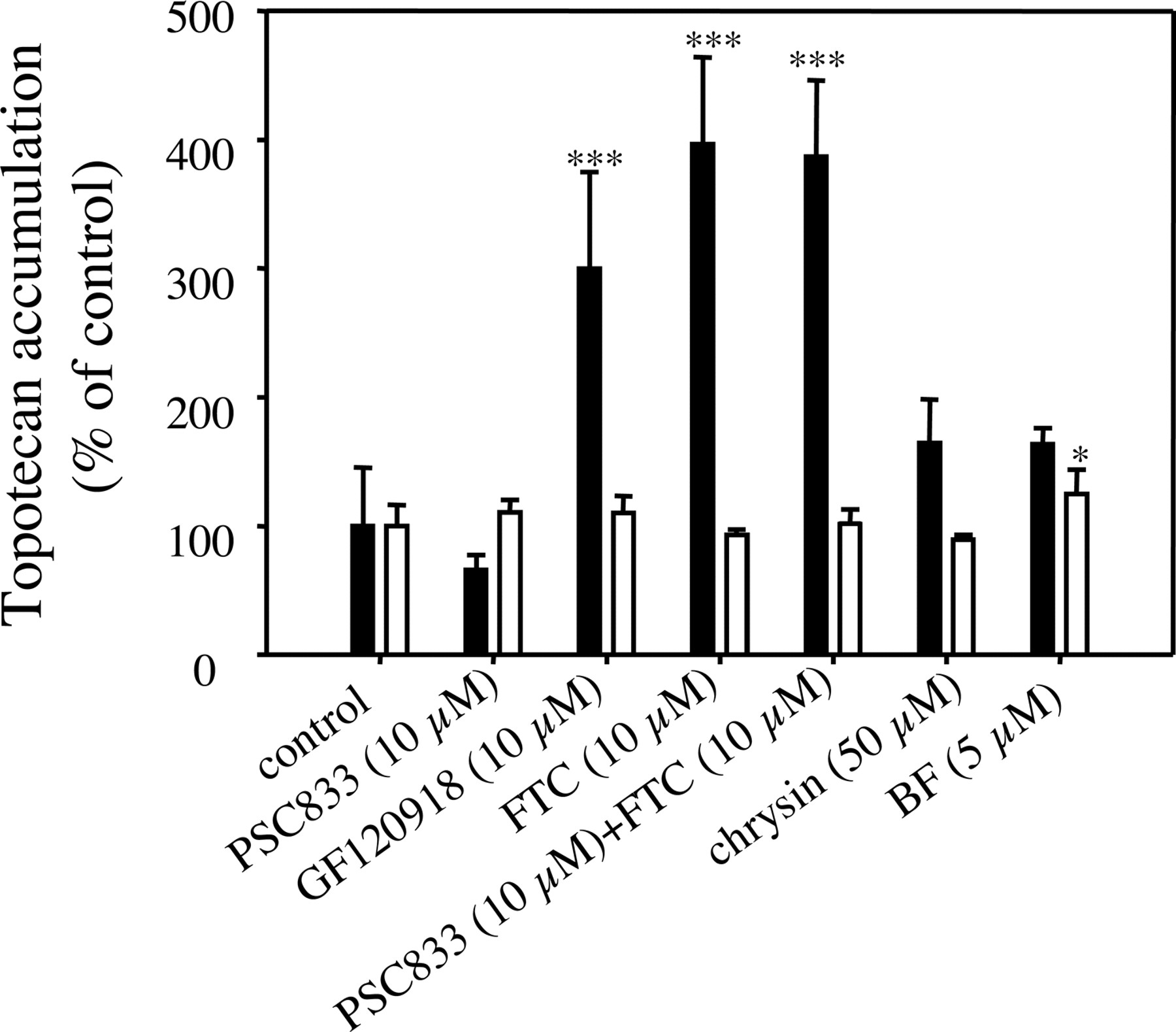
Topotecan Accumulation in MDCK-Mock and MDCK-Bcrp1 Cells. To understand the interaction of chrysin and BF with mouse Bcrp1-mediated transport of topotecan, we characterized the effects of these flavonoids on topotecan accumulation in both MDCK-mock and MDCK-Bcrp1 cells (MDCK cells transfected with mouse Bcrp1). As shown in Fig. 4, none of the tested compounds at specified concentrations significantly increased topotecan accumulation in MDCK-mock cells except that BF (5 μM) slightly increased topotecan accumulation in these cells (125 ± 18.9% of control, p < 0.05). In MDCK-Bcrp1 cells, both FTC (10 μM), a specific BCRP inhibitor (Rabindran et al., 1998), and GF120918 (10 μM), an inhibitor of both BCRP and P-glycoprotein (Allen et al., 1999), significantly increased topotecan accumulation (396 ± 67.6% and 300 ± 75.0% for FTC and GF120918, respectively, p < 0.001 for both compounds); however, PSC833 (10 μM), a potent P-glycoprotein inhibitor with negligible effect on BCRP (Bcrp1)-mediated transport (Jonker et al., 2000), had no significant effect on topotecan accumulation in these cells (65.8 ± 11.8%, p > 0.05). The topotecan accumulation in the presence of a combination of 10 μM FTC and 10 μM PSC833 (387 ± 59.4%) was almost the same as that in the presence of 10 μM FTC alone (396 ± 67.6%). All these results indicate that the accumulation of topotecan in MDCK-Bcrp1 cells is limited predominantly by the expression of mouse Bcrp1 in these cells. The flavonoids chrysin (50 μM) and BF (5 μM) only slightly increased (p > 0.05) topotecan accumulation in MDCK-Bcrp1 cells (164 ± 34.0% and 163 ± 12.9% for chrysin and BF, respectively), and the average increase of topotecan accumulation by chrysin (50 μM) and BF (5 μM) was only 22% and 21% of that produced by 10 μM FTC, respectively, indicating that chrysin and BF have only weak, if any, inhibition activity against mouse Bcrp1 transfected in MDCK cells.

Effects of chrysin and BF on topotecan accumulation in MDCK-mock and MDCK-Bcrp1 cells. The 10-min accumulation of topotecan (5 μM) in MDCK-mock (open bars) and MDCK-Bcrp1 cells (black bars) in the presence of 0.1% DMSO (control) or the test compounds at specified concentrations was determined as described under Materials and Methods. The accumulation of topotecan was expressed as percentage of the control accumulation. Data are expressed as mean ± S.D., n = 4. ★★★, p < 0.001.
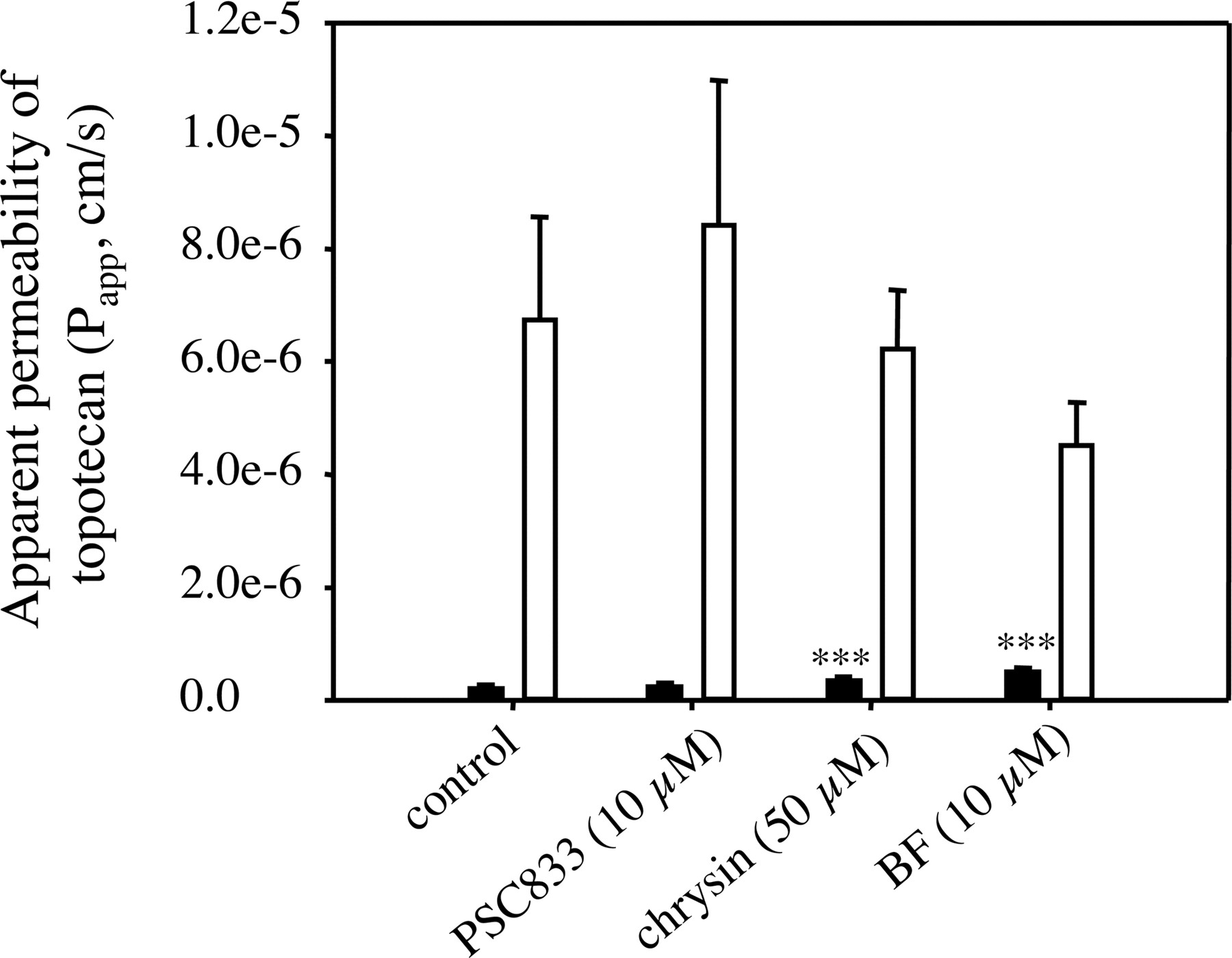
Bidirectional Transport of Topotecan across MDCK-Bcrp1 Cell Monolayers. To further confirm the observations we obtained in the study of topotecan accumulation in MDCK cells, we also evaluated the effects of the flavonoids chrysin and BF on topotecan bidirectional transport across MDCK-Bcrp1 cells. It has been demonstrated that Bcrp1 is expressed in the apical membrane of MDCK-Bcrp1 cells (Jonker et al., 2000). As we can see from Fig. 5, the Papp,BL-AP of topotecan across MDCK-Bcrp1 cells (6.75 ± 1.81 × 10–6 cm/s) was consistently much higher (p < 0.001) than the Papp,AP-BL of topotecan (0.208 ± 0.0710 × 10–6 cm/s), with a transport ratio of 32.6. This polarized transport was not due to the low level of intrinsic P-glycoprotein expression in MDCK cells, because it was not significantly affected by 10 μM PSC833 (control versus PSC833 for topotecan Papp,BL-AP, 6.75 ± 1.81 × 10–6 cm/s versus 8.43 ± 2.56 × 10–6 cm/s, p > 0.05; control versus PSC833 for topotecan Papp,AP-BL value, 0.208 ± 0.0710 × 10–6 cm/s versus 0.243 ± 0.0651 × 10–6 cm/s, p > 0.05) (Fig. 5). The flavonoids chrysin (50 μM) and BF (10 μM) only slightly increased topotecan Papp,AP-BL [0.208 ± 0.0710 × 10–6 cm/s, 0.350 ± 0.0768 × 10–6 cm/s (p < 0.001), and 0.504 ± 0.0737 × 10–6 cm/s (p < 0.001) for control, chrysin, and BF, respectively] and slightly decreased the Papp,BL-AP [6.75 ± 1.81 × 10–6 cm/s, 6.24 ± 1.03 × 10–6 cm/s (p > 0.05), and 4.52 ± 0.756 × 10–6 cm/s (p > 0.05) for control, chrysin, and BF, respectively) of topotecan (Fig. 5), indicating that the flavonoids chrysin and BF, indeed, only have weak inhibition activity against mouse Bcrp1 in MDCK cells.

Apparent permeability of topotecan (Papp) across MDCK-Bcrp1 cell monolayers. The apparent permeability of topotecan across MDCK-Bcrp1 cell monolayers in both AP-to-BL (black bar) and BL-to-AP (open bar) directions in the presence of specified concentrations of the test compounds or 0.1% DMSO (control) (applied in both apical and basolateral chambers) was determined as described under Materials and Methods. Topotecan (10 μM) was added to either the apical (AP-to-BL) or basolateral (BL-to-AP) chamber following a 30-min preincubation of the cell monolayers with the test compounds (or 0.1% DMSO). The samples (150 μl) were taken from the opposite chamber at 30, 60, and 90 min after addition of topotecan and replaced with the same volume of the fresh transport buffer. Data are expressed as mean ± S.D., n = 3 or 6. ★★★, p < 0.001 (compared with the corresponding control value).
Discussion
The objective of the present study was to determine whether the flavonoid-BCRP interactions observed in previous in vitro studies (Cooray et al., 2004; Imai et al., 2004; Zhang et al., 2004b) can be translated into in vivo pharmacokinetic interactions. These in vitro-in vivo association studies are very important because the recent resurgence of scientific interest in flavonoids has resulted in sharply increasing consumption of these compounds as dietary supplements in the general population, raising a serious concern regarding their potential interactions with conventional medications. To predict these potential interactions, in vitro mechanistic studies, as well as in vitro-in vivo association studies, are necessary to provide valuable information regarding interactions.
The flavonoids selected for this study are chrysin and BF. Both compounds have been shown to be potent BCRP inhibitors, with their EC50 values for inhibiting BCRP-mediated transport of mitoxantrone (a model BCRP substrate) in MCF-7 MX100 cells less than 1 μM. Since topotecan was used as the model BCRP substrate for the in vivo pharmacokinetic studies in this report, we first investigated the effects of these flavonoids on BCRP-mediated transport of topotecan. It was demonstrated that both chrysin (50 μM) and BF (5 μM) could significantly increase topotecan accumulation in BCRP-overexpressing MCF-7 MX100 cells without any significant effect in BCRP-negative MCF-7/sensitive cells. Because MCF-7 MX100 cells have been shown to have little P-glycoprotein, MRP1, or MRP2 expression (Zhang et al., 2004a,b), and topotecan accumulation in these cells can be significantly increased by FTC but not PSC833, we concluded that the flavonoids chrysin and BF can inhibit BCRP-mediated transport of topotecan. This conclusion is also consistent with a recent report (Imai et al., 2004), in which the flavonoids genistein and naringenin were also shown to inhibit BCRP-mediated efflux of topotecan. Furthermore, the net increases of topotecan accumulation produced by 50 μM chrysin and 5 μM BF reached 119% and 78% of that produced by 10 μM FTC in MCF-7 MX100 cells. Since we have shown previously that FTC at a 10 μM concentration can completely inhibit BCRP in MCF-7 MX100 cells (Zhang et al., 2004a), nearly complete inhibition of BCRP-mediated transport of topotecan may be attainable with 50 μM chrysin or 5 μM BF.
The pharmacokinetic studies in this report were mainly conducted in SD rats. Although it has been demonstrated that topotecan bioavailability and biliary excretion are markedly affected by the expression of BCRP in mdr1a/1b (–/–) mice and in humans (Jonker et al., 2000; Kruijtzer et al., 2002), similar observations in rats have not been experimentally demonstrated. Therefore, to confirm that the bioavailability of topotecan is also limited by the expression of BCRP in rats, we compared the pharmacokinetics of topotecan after oral administration of topotecan with and without GF120918 in SD rats. The absolute bioavailability of topotecan without GF120918 coadministration was shown to be 29.7 ± 14.8%, which is similar to the bioavailability of topotecan in humans (30–40%) (Kruijtzer et al., 2002). As expected, GF120918 significantly increased the AUC and bioavailability of topotecan by more than 4-fold. Although GF120918 is a potent inhibitor of both BCRP and P-glycoprotein, we believe that this increase of topotecan AUC and bioavailability by GF120918 is mainly due to the inhibition of rat BCRP instead of P-glycoprotein, since topotecan has been shown to be a very good BCRP substrate but only a moderate or poor P-glycoprotein substrate. In mdr1a/1b (–/–) mice, which do not have any functional P-glycoprotein expression, GF120918 can increase topotecan AUC by more than 6-fold after oral topotecan administration; however, the AUC of topotecan in mdr1a/1b (–/–) mice after oral topotecan administration was only about 2-fold that in the wild-type mice (Jonker et al., 2000). The apparent topotecan bioavailability value of greater than 100% (130 ± 58.8%) after oral coadministration of topotecan with GF120918 in SD rats also indicates that the increase of the topotecan AUC resulted not only from the increased intestinal absorption but also from the decreased clearance, consistent with the mdr1a/1b (–/–) mouse study (Jonker et al., 2000).
Although we have shown that chrysin can inhibit BCRP-mediated transport of topotecan with high potency, and inhibition of BCRP can markedly increase the AUC and bioavailability of topotecan in SD rats, oral coadministration of topotecan with chrysin did not increase the AUC and bioavailability of topotecan in rats. This lack of in vitro-in vivo association was also observed in mdr1a/1b (–/–) mice. One possible reason for this inconsistency might be due to the metabolism of chrysin in the intestine. Chrysin has two hydroxyl groups and does undergo extensive glucuronidation and sulfation (Galijatovic et al., 1999); thus, it is possible that, due to the extensive metabolism of chrysin in the intestine, the intestinal concentration of this compound is not high enough to inhibit BCRP. To test this possibility, we next conducted a study to look at the effects of BF on topotecan pharmacokinetics in SD rats. BF has higher BCRP inhibition activity than chrysin, and the molecule does not have any hydroxyl groups, and thus the parent compound does not undergo rapid conjugative metabolism. However, coadministration of BF still did not have any significant effect on topotecan pharmacokinetics, indicating that metabolism may be not the main reason, or at least not the only reason, for this lack of in vitro-in vivo association. This supposition is also supported by the fact that the flavonoid quercetin, which has five hydroxyl groups and undergoes rapid conjugative metabolism (Murota et al., 2000), can still significantly increase paclitaxel bioavailability in rats, presumably by inhibition of P-glycoprotein and CYP3A (Choi et al., 2004b). Therefore, extensive intestinal metabolism may not represent the main reason for the observed in vitro-in vivo discrepancy.
Another possibility that might result in the observed in vitro-in vivo inconsistency is related to the potential species differences with regard to flavonoid-BCRP interactions. The in vitro inhibition of BCRP-mediated transport of topotecan by the flavonoids chrysin and BF was observed in human breast cancer MCF-7 MX100 cells; however, the in vivo topotecan pharmacokinetic studies were performed in rats or mice. Mouse Bcrp1 shares only 81% amino acid identity with human BCRP (Allen et al., 1999), and even a single amino acid mutation at position 482 of BCRP was shown to significantly alter BCRP substrate and antagonist specificity (Robey et al., 2003). To test the possibility of a species difference in inhibitor potency, we evaluated the interactions of chrysin and BF with mouse Bcrp1-mediated transport of topotecan by determining the effects of these compounds on topotecan accumulation in, and bidirectional transport across, MDCK-Bcrp1 (MDCK cells transfected with mouse Bcrp1) cells. PSC833, the P-glycoprotein inhibitor, had no significant effect on topotecan accumulation in either MDCK-Bcrp1 or MDCK-mock cells at a concentration (10 μM) high enough to inhibit P-glycoprotein (Achira et al., 1999), indicating no P-glycoprotein-mediated transport of topotecan in these cells. The flavonoids chrysin (50 μM) and BF (5 μM) only slightly and insignificantly increased topotecan accumulation in MDCK-Bcrp1 cells, and the net increases of topotecan accumulation produced by these compounds were only about 20% of that produced by 10 μM FTC. Thus, chrysin and BF may indeed have much lower inhibition activity against mouse Bcrp1 than against human BCRP. This lack of potent inhibition of mouse Bcrp1 was also confirmed by the topotecan bidirectional transport study using the MDCK-Bcrp1 cell monolayers. Therefore, the lack of significant pharmacokinetic interaction of the flavonoids chrysin and BF with topotecan may be due, at least in part, to their lack of potent inhibition of rat and mouse BCRP. It could be argued that the lack of potent mouse Bcrp1 inhibition by chrysin and BF might be due to the extensive metabolism of the flavonoids in MDCK-Bcrp1 cells instead of an intrinsically weaker inhibition activity. Although this explanation could not be totally ruled out by the data presented in this study, we think it is less likely, based on the following considerations. First of all, MDCK cells, derived from dog renal distal tubular cells, do not exhibit substantial expression of drug-metabolizing enzymes (Lohr et al., 1998). Second, although chrysin has two hydroxyl groups and does undergo extensive glucuronidation and sulfation (no oxidation) (Galijatovic et al., 1999), the metabolism of this compound (20 μM) in Caco-2 cells was minimal following a 1-h incubation (Walle et al., 1999), and genistein (which contains three hydroxyl groups), a flavonoid with a structure similar to that of chrysin, was shown to be metabolically stable for about 4 h when incubated with renal proximal tubular LLC-PK1 cells (Imai et al., 2004). Third, the BF molecule has no hydroxyl groups at all, and therefore the parent compound does not undergo conjugative metabolism. Lastly, our topotecan accumulation study was performed following only a 10 min-incubation. Therefore, it may be unlikely that the lack of potent Bcrp1 inhibition by chrysin and BF was mainly due to metabolism of the flavonoids. However, to completely rule out this possibility and other potential confounding factors, the kinetic parameters of chrysin and BF for inhibiting human BCRP and mouse Bcrp1 expressed (transfected) in the same cell line have to be carefully evaluated and compared. These studies are now ongoing in our laboratory.
Other possible explanations for the observed in vitro-in vivo discrepancy could not be excluded. For example, chrysin and BF may also inhibit some uptake transporter of topotecan in addition to BCRP and in this manner negate the effect of BCRP inhibition on topotecan AUC and bioavailability. Topotecan has an α-hydroxy-δ-lactone ring, which can undergo reversible pH-dependent hydrolysis, producing a ring-open carboxylate form. At physiological pH values, the carboxylate form predominates, whereas under acidic pH, the lactone form predominates (Underberg et al., 1990; Dennis et al., 1997). The carboxylate form is expected to be ionized at physiological pH and, therefore, cannot pass through intestinal epithelial cells by simple diffusion. However, whether some organic anion transporters (such as organic anion-transporting polypeptide) are involved in the intestinal absorption of the ionized species is currently unknown. It has been reported that coadministration of probenecid with topotecan can increase the AUC and decrease the systemic clearance of topotecan hydroxy acid (Zamboni et al., 1998), indicating a possible involvement of one or multiple transporters in the uptake of topotecan hydroxy acid into cells. Flavonoids have been shown to inhibit some organic anion transporters such as organic anion-transporting polypeptide (Dresser et al., 2002) and MRP1 (Nguyen et al., 2003). Therefore, it cannot be excluded that the observed in vivo-in vitro discrepancy might be due to the inhibition of some intestinal uptake transporters of topotecan by flavonoids, which negated the effect of BCRP inhibition on topotecan pharmacokinetics.
In conclusion, we have demonstrated in this study that the flavonoids chrysin and BF can inhibit human BCRP-mediated transport of topotecan with relatively high potency. We also demonstrated for the first time that GF120918 can significantly increase the AUC and bioavailability of topotecan in rats by more than 4-fold after oral coadministration, and this increase of AUC and bioavailability is mainly due to the inhibition of rat BCRP. However, oral coadministration of topotecan with the potent BCRP inhibitors chrysin and BF failed to significantly alter topotecan pharmacokinetics in rats or mdr1a/1b (–/–) mice. The reason(s) for this lack of in vitro-in vivo association may be due to the fact that chrysin and BF only potently inhibit topotecan efflux by human BCRP, but not rat or mouse BCRP. However, this interspecies difference needs further confirmation in studies using the same cell line transfected with BCRP from different species. In addition, the intestinal metabolism of flavonoids, as well as the potential inhibition of other transporters by flavonoids, may also contribute to the lack of in vivo effect observed in the present investigation.
Acknowledgments
We thank Kosea Frederick for technical assistance with the liquid chromatography-tandem mass spectrometry assays.
Footnotes
-
Financial support for this study was provided by grants from the Susan G. Komen Breast Cancer Foundation and Pfizer Global Research and Development. S.Z. was the recipient of a graduate fellowship from Pfizer Global Research and Development.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002501.
-
ABBREVIATIONS: BCRP, breast cancer resistance protein; BF, 7,8-benzoflavone; FTC, fumitremorgin C; GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide; MCF-7 MX100, MCF-7 cells selected with mitoxantrone; MRP1, multidrug resistance-associated protein 1; PBS, phosphate-buffered saline; HBSS, Hanks' buffered salt solution; PSC833, valspodar; HPLC, high-performance liquid chromatography; DMSO, dimethyl sulfoxide; SD, Sprague-Dawley; AP, apical; BL, basolateral; AUC, area under the plasma concentration-time curve.
- Received September 26, 2004.
- Accepted December 16, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}