


Abstract
The enzymic basis for intracellular reduction of N-hydroxylated amidines to their corresponding amidines, and hydroxylamines to their corresponding amines, is unknown. The hydroxylated amidines can be used as prodrug moieties, and an understanding of the enzyme system active in the reduction can contribute to more efficient drug development. In this study, we examined the properties of this enzyme system using benzamidoxime and N-hydroxymelagatran as substrates. In rats and humans, the hepatic enzyme system was localized in mitochondria as well as in microsomes, using preferably NADH as cofactor. Potassium cyanide, N-methylhydroxylamine, p-hydroxymercuribenzoate, and desferrioxamine were efficient inhibitors, whereas typical cytochrome P450 (P450) inhibitors were ineffective. In rats, the highest specific activity was found in liver, adipose tissue, and kidneys, whereas in humans, the specific activity in the preparations of adipose tissue examined was lower. A sex difference was observed in rat liver, where 4-fold higher activity was seen in microsomes from female rats. No gender differences were present in any other tissue investigated. Partial purification of the hepatic system was achieved using polyethylene glycol fractionation followed by Octyl Sepharose chromatography at low detergent concentrations, whereas the enzyme was denatured after complete solubilization. The unique appearance of the enzyme activity in adipose tissue, together with the cyanide sensitivity and the failure of typical P450 inhibitors to impede the reaction, indicates that the enzyme system active in reduction of benzamidoxime and N-hydroxymelagatran formation is not of cytochrome P450 origin, but likely consists of an NADH-dependent electron transfer chain with a cyanide-sensitive protein as the terminal component.
The reduction of N-hydroxylated amidines (amidoximes) is of great relevance in the prodrug concept, where amidoxime groups serve as prodrug moieties for amidino groups. The reduction of N-hydroxylated amines is probably catalyzed by the same enzyme system, as evidenced by their similar properties as revealed from the literature, and this reaction is important in the detoxification of many xenobiotics, as well as metabolism of pharmaceutical drugs (King et al., 1999; Clement, 2002). The enzyme system responsible for these reactions is unknown, but its identification would be of great importance for understanding the metabolism and disposition of drugs and other xenobiotic compounds.
The features of a membrane-bound mammalian mitochondrial hydroxylamine reductase were first described some 37 years ago (Bernheim and Hochstein, 1968), and the system was further characterized during subsequent years (Bernheim, 1969, 1972). The reductase system has been shown to be responsible for the N-reduction of other aliphatic hydroxylamines such as N-methylhydroxylamine, as well as the clinically relevant detoxification process of the hydroxylamines of methamphetamine and amphetamine (Clement et al., 2000).
Sulfamethoxazole, a sulfonamide antimicrobial, can undergo N-hydroxylation by a cytochrome P450 enzyme (CYP2C9) to form sulfamethoxazole hydroxylamine (Cribb et al., 1995), which, in turn, is spontaneously converted to the nitroso metabolite. Both these compounds are associated with idiosyncratic delayed drug hypersensitivity, and in vitro studies have supported covalent protein binding, thereby possibly triggering an idiosyncratic reaction (Cribb et al., 1996). The sulfamethoxazole hydroxylamine can be reduced back to the parent sulfonamide by a N-hydroxylamine reductase system (Cribb et al., 1995; Trepanier and Miller, 2000), which is therefore of great importance for detoxification.
Yet another example of an important function of the N-hydroxylamine reductase is the formation of melagatran, the active form of the novel, oral direct thrombin inhibitor ximelagatran (Exanta; AstraZeneca) (Gustafsson et al., 2001, 2004; Gustafsson and Elg, 2003). Melagatran is a potent competitive inhibitor of the human serine protease α-thrombin (Gustafsson et al., 1998). The prodrug ximelagatran contains two groups that protect the hydrophilic and ionized moieties of melagatran: an ethyl group on the carboxylic acid and a hydroxy group on the amidine, yielding, as a consequence, good oral absorption of the compound. Formation of melagatran requires the hydrolysis of the ester and the reduction of the N-hydroxyamidine (Fig. 1), the latter reduction most likely carried out by the N-hydroxylamine reductase system discussed above.

Structures of putative substrates and products as a consequence of reduction by the enzyme system studied. A, reduction of benzamidoxime to benzamidine; B, two-step bioactivation of the double prodrug ximelagatran, via two different pathways and different intermediates, to melagatran; C, reduction of N-methylhydroxylamine to N-methylamine; and D, reduction of sulfamethoxazole hydroxylamine to sulfamethoxazole.
The origin of the N-hydroxylamine reductase system was suggested, based on reconstitution experiments with partially purified isoenzyme components from pig liver, to consist of cytochrome b5, its reductase, and a third microsomal protein (Kadlubar and Ziegler, 1974). Clement et al. (1997) have suggested that the third component, using benzamidoxime as a substrate, would consist of a cytochrome P450 isoenzyme of subfamily 2D. In this study, we have characterized and partially purified the enzyme system active in the reduction of benzamidoxime and N-hydroxymelagatran from both human and rat liver microsomes and mitochondria.
Materials and Methods
N-Hydroxymelagatran and melagatran, as well as deuterium and 13C-labeled melagatran (internal standard, isotopic purity >99.9%), were supplied by AstraZeneca R&D Mölndal (Mölndal, Sweden). The following substrates and chemical compounds were purchased from the companies indicated: benzamidoxime (Tokyo Kasei Kogyo Co. Ltd., Tokyo, Japan); benzamidine, dextromethorphan, HEPES, ketoconazole, p-hydroxymercuribenzoic acid, methimazole, metyrapone, NADH, NADPH, N-ethylmaleimide, 1-octane sulfonic acid, and 1,10-phenantroline monohydrate (Sigma-Aldrich, St. Louis, MO); CO (Air Liquide, Malmö, Sweden); desferrioxamine (Novartis Ltd., Basel, Switzerland); KCN and N-methyl-hydroxylamine hydrochloride (Aldrich Chemie GmbH, Steinheim, Germany); and PEG 6000 (Merck, Briare le Canal, France). SKF525A was a kind gift from GlaxoSmithKline (Welwyn Garden City, Hertfordshire, UK). Human liver samples were obtained from Sahlgrenska Hospital (Gothenburg, Sweden) and originated from patients undergoing liver resection. All tissues were obtained through qualified medical staff, with donor consent and with the approval of the local ethics committee at Sahlgrenska Hospital. Human adipose tissue was bought from Pathlore Biomaterial Resource (Nottingham, UK), and human lung, jejunum, and kidney microsomes were purchased from In Vitro Technologies (Baltimore, MD). Rats were of the Sprague-Dawley strain and bought from Scanbur BK AB (Sollentuna, Sweden). Ethical permission was obtained for all samples.
Preparation of Tissue Samples. Rats were killed with CO2, and their organs were excised, washed, weighed, and put in ice-cold 0.9% (w/v) NaCl solution. Frozen tissue pieces from various human organs were ground to a powder in a mortar in liquid nitrogen.
Isolation of Mitochondrial and Microsomal Fractions Used for in Vitro Incubations. Tissue samples were homogenized in mitochondria buffer [10 mM Tris-acetate, pH 7.4 (4°C), 0.25 M sucrose, and 0.5 mM EDTA] to a 10 to 35% (w/v) homogenate. Livers and kidneys were homogenized either using a smooth Teflon pestle tissue grinder with approximately four strokes at 1650 rpm or minced. The latter method was also used for lung, jejunum, duodenum, and adipose tissue.
Mitochondria were prepared by the method originally described by Wikvall (1984), here denoted protocol A, with some minor modifications. In brief, the homogenate was centrifuged at 800g for 10 min, the pellet discarded, and the supernatant centrifuged at 6500g for 20 min to pellet the mitochondria, which were further washed another two or three times in mitochondria buffer before finally being resuspended in it. Subsequently, the 6500g supernatant was centrifuged at 100,000g for 1 h to pellet the microsomes. After suspension in 1 to 2 volumes of 10 mM sodium/potassium phosphate, pH 7.4 (4°C), containing 1.14% (w/v) KCl, the microsomes were again centrifuged before finally being resuspended in 50 mM potassium phosphate buffer, pH 7.4. The microsomes were stored in aliquots at -70°C.
Isolation of Mitochondrial and Microsomal Fractions Used for Partial Enzyme Purifications. The tissue samples were homogenized to a 25% (w/v) homogenate in H-buffer [220 mM d-mannitol, 70 mM sucrose, 2 mM HEPES, pH 7.4 (4°C)] or in 10 mM sodium/potassium phosphate buffer, pH 7.4 (4°C), containing 1.14% (w/v) KCl and 0.25 mM PMSF (the latter was used when only microsomes were isolated).
Mitochondria were prepared by a method for high-yield preparation of mitochondria, here denoted protocol B, first described by Bustamante et al. (1977), and slightly modified as in Pedersen et al. (1978). In brief, the homogenate was centrifuged at 1100g for 3 min and the pellet washed three times in 1 volume of H-buffer, with the supernatants saved in between. The foremost resulting supernatant was centrifuged at 6500g for 20 min, and the following three supernatants were pooled and centrifuged at 6750g for 15 min to pellet the mitochondria. The mitochondrial pellet was washed twice by resuspending in half the previous volume and recentrifuged at 20,000g for 10 and 15 min, respectively. This final two-layered mitochondrial pellet, with a more reddish and softer upper layer (here denoted mitochondria fluff) and a more brownish, harder lower layer, was resuspended in layers in H-buffer. The 6500g supernatant was further subjected to centrifugation at 100,000g for 1 h to pellet the microsomes, which were resuspended in 1 to 2 volumes of 0.1 M potassium pyrophosphate buffer containing 0.25 mM PMSF, and recentrifuged before final resuspension in A-buffer [10 mM Tris-HCl, pH 7.4, containing 20% (w/v) glycerol and 1 mM EDTA] with 0.25 mM PMSF.
Subfractionation of Rat Liver Mitochondria. Rat liver mitochondria were prepared by protocol A, with the exception that the last two resuspensions were made in 0.25 M sucrose. To retrieve pure mitochondrial outer membrane, the mitochondria were subjected to fractionation by digitonin treatment (Schnaitman et al., 1967; Schnaitman and Greenawalt, 1968) and sucrose gradient according to the method of Benga et al. (1979), with slight modifications. Briefly, mitochondrial suspension (40 mg/ml) was incubated with an equal volume of ice-cold digitonin solution (6 mg/ml in 0.25 M sucrose) on ice with continuous stirring for 20 min, followed by centrifugation at 9500g for 15 min to pellet the mitoplasts. The supernatant was further centrifuged at 100,000g for 1 h, and the pelleted crude outer membrane fraction was resuspended in 0.25 M sucrose in 10 mM Tris-HCl (pH 7.4) and frozen at -70°C until use. The thawed crude outer membrane solution was diluted to a protein concentration of 7 mg/ml, and 2.8 ml was laid on top of a discontinuous sucrose gradient. The gradient consisted of 2.4 ml of 23.2% sucrose, 2.4 ml of 37.4% sucrose, and 3.6 ml of 51.3% sucrose, all in 20 mM phosphate buffer, pH 7.4, and was further centrifuged at 97,000g for 2 h. Ten fractions were removed from the top of the tube, with fraction 1 corresponding to the uppermost one.
Protein Concentration Determination. Protein concentration was measured according to the method of Lowry et al. (1951), using bovine serum albumin as standard.
Assay for Reduction of Benzamidoxime to Benzamidine. The incubation mixture consisted of 0.2 to 0.7 mg/ml mitochondrial or microsomal protein from different human or rat tissues and 0.5 mM benzamidoxime, in 50 to 100 mM potassium phosphate buffer, pH 6.3. The samples were preincubated for 3 to 5 min at 37°C, and the catalytic reaction was initiated by addition of 250 μM or 1 mM NADH, in a total incubation volume of 60 to 300 μl. After 20 or 30 min at 37°C, the reaction was terminated by addition of 1 volume of ice-cold methanol, briefly mixed, and cooled on ice. Samples were then centrifuged on maximal speed in a 4°C microcentrifuge for >5 min to pellet the precipitate. Supernatants were subjected to HPLC for further analysis.
The effect of CO on the benzamidoxime reduction was analyzed after 1 to 2 min of bubbling of human liver microsomes with CO. Control incubations were bubbled with nitrogen.
HPLC Detection of the Reduction of Benzamidoxime to Benzamidine. The formation of benzamidine in the in vitro incubations was assessed using a HPLC system consisting of a Varian ProStar model 410 autosampler, Varian ProStar model 240 solvent delivery module, and a Varian ProStar 310UV-visible detector with Star Chromatography Workstation version 5.51 software (all from Varian, Inc., Palo Alto, CA). All other equipment and settings were as stated before (Clement et al., 1997), with the exception of using an isocratically distributed mobile phase of 10 mM, instead of 3 mM, 1-octane-sulfonic acid (pH adjusted to 2.5 with 85% phosphoric acid), mixed with 12% acetonitrile. Sample aliquots of 20 to 30 μl were injected.
Assay for Reduction of N-Hydroxymelagatran to Melagatran. The incubation mixture comprised 0.4 to 0.5 mg/ml mitochondrial or microsomal protein from different human tissues, 1 mM N-hydroxymelagatran, in 100 mM potassium phosphate buffer, pH 6.3. Samples were preincubated for 2 min at 37°C; the reaction was initiated by addition of 250 μM NADH and terminated after 20 min by the addition of formic acid to a concentration of 0.5 M. Samples were then centrifuged at 4°C in a microcentrifuge at maximum speed for >5 min or at 4000g for 20 min to pellet precipitate. Of the resulting supernatant, 40 μl was mixed with 160 μl of internal standard, deuterium, and 13C-labeled melagatran (solved in mobile phase), to a final concentration of 0.4 μM. Thereafter, 10 μl was injected for analysis by LC-MS.
LC-MS Detection of the Reduction of N-Hydroxymelagatran to Melagatran. The LC system consisted of an HP 1100 series LC pump and column oven (Agilent Technologies Deutschland, Waldbronn, Germany), combined with an HTS PAL injector (CTC Analytics, Zwingen, Switzerland). LC separations were performed on a reversed-phase HyPURITY C18 analytical column (100 mm × 2.1 mm i.d., 5 μm; ThermoQuest, Runcorn, UK) at 40°C. The mobile phase consisted of 10% acetonitrile, 5 μM acetic acid, and 10 mM ammonium acetate, resulting in an apparent pH ∼5. An isocratic elution was used at 0.75 ml/min. The retention times of melagatran, internal standard, and N-hydroxymelagatran were 1.3, 1.3, and 2.2 min, respectively. Detection was performed with a triple quadrupole mass spectrometer, API4000, equipped with electrospray interface (Applied Biosystems/MDS Sciex, Foster City, CA). The mass spectrometer operated at turbo heater temperature, at 550°C, nebulizer gas (GS1) 60 pounds per square inch gauge (psig), turbo gas (GS2) 70 psig, curtain gas 30 psig, electrospray voltage 4 kV, in positive mode. The orifice voltage was set at 81 V, collision energy at 33 V, and collision-activated dissociation gas at 10 psig. The multiple reaction monitoring transitions of the precursor ions (M + H)+ to the corresponding product ions were m/z 430.1 to 233.3 for melagatran and m/z 434.1 to 233.3 for its isotope-labeled internal standard. A dwell time of 200 ms was used for melagatran and 100 ms for the internal standards. Instrument control, data acquisition, and data evaluation were performed using Applied Biosystems/MDS Sciex Analyst 1.4 software.
Western Blot Analysis. Denatured protein samples (20 μg of protein/sample) were resolved by 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Hybond-C; Amersham Biosciences AB, Uppsala, Sweden) by wet blotting. For immunostainings, the following specific antibodies were used: polyclonal rabbit anti-ERp29 as microsomal marker (1:1000; kindly provided by Dr. S. Mkrtchian, Division of Molecular Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Sweden), polyclonal rabbit anti-VDAC1/porin for outer membrane detection (1: 2000; Sigma Chemie, Deisenhofen, Germany), and monoclonal anti-Tim23 for mitochondria inner membrane (1:2500; BD Biosciences); horseradish peroxidase-conjugated goat antibody to mouse IgG (1:1500; DakoCytomation Denmark A/S, Glostrup, Denmark); or horseradish peroxidase-conjugated goat antibody to rabbit IgG (1:2000; DakoCytomation Denmark A/S). Signals were detected with chemiluminescence reagent (SuperSignal West Pico; Pierce Chemical, Rockford IL), according to the manufacturer's protocol.
Partial Enzyme Purification.PEG 6000 Precipitation. Human- and rat-derived liver microsomes, as well as mitochondria originating from the reddish, soft layer (mitochondria fluff) prepared according to protocol B, described above, were diluted to a concentration of approximately 20 mg/ml in A-buffer (10 mM Tris-HCl, pH 7.4, 20% glycerol, and 1 mM EDTA) containing 0.25 mM PMSF. Another 5 volumes of B-buffer (100 mM Tris-HCl, pH 7.4, 100 mM KCl, 20% glycerol, and 1 mM EDTA) containing 0.25 mM PMSF were added. Samples were bubbled with nitrogen gas for 1 min, fresh sodium cholate solution, 10% (w/v), was added to a final concentration of 1.7% (w/v), and the solution was stirred for 20 to 30 min. A 50% (w/v) PEG 6000 solution was added drop by drop to the protein solution to a final concentration of 6% (w/v) PEG and stirred for 15 min before centrifugation at 5000g for 10 min at 4°C. Decanted supernatant was further precipitated by PEG 6000 in increments of 3% until it contained 21%. Precipitated protein pellets were resuspended and dialyzed for >15 h in A-buffer using a 12,000 to 14,000 MWCO Spectra/Por membrane (Spectrum Laboratories, Inc., Rancho Dominguez, CA). Catalytic activity was assessed for all the dialyzed fractions using the benzamidoxime to benzamidine reduction assay, and active fractions were further subjected to Octyl Sepharose chromatography.
Octyl Sepharose Chromatography. The PEG fractions containing N-hydroxylamine reductase activity were further chromatographed on 1-ml columns of 1 ml of Octyl Sepharose 4 Fast Flow (Amersham Biosciences AB). Columns were equilibrated with 200 mM sodium phosphate buffer, pH 6.8, containing 10% (w/v) glycerol, 1 mM EDTA, and 0.75% (v/v) Tween 20. The sample mix, consisting of 0.25 ml of dialyzed PEG fraction (corresponding to 2.5–6 mg of protein), 62.5 μl of 1 M potassium phosphate buffer, pH 6.3, and 35 μl of 10% Tween 20 was applied on the column and eluted with equilibration buffer, collecting 25 fractions of 67 μl. For each fraction, catalytic activity was determined using the benzamidoxime to benzamidine reduction assay.
Results
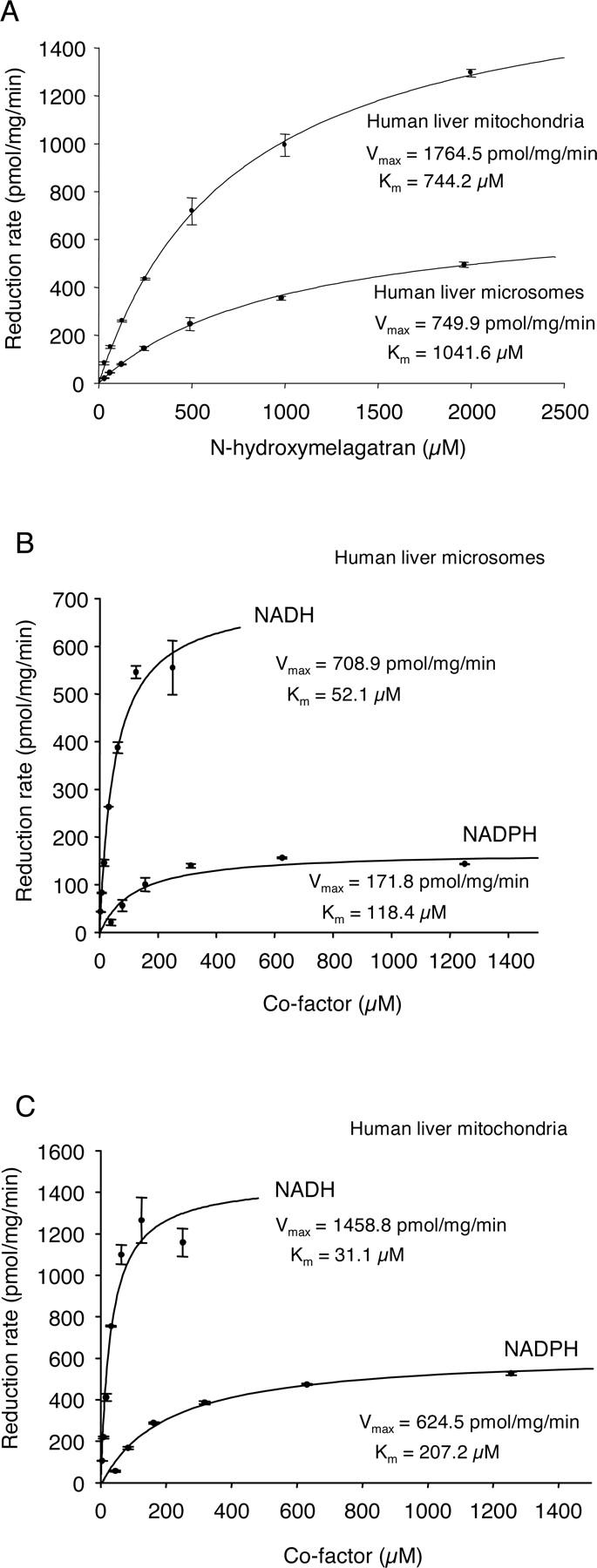
N-Reduction of N-Hydroxylated Amidines. To characterize the enzyme system active in the reduction of amidoximes, we studied the in vitro reduction of benzamidoxime to benzamidine (Fig. 1A), as well as the reduction of N-hydroxymelagatran to melagatran (Fig. 1B). The reduction of N-hydroxymelagatran was shown to exert a pH optimum of 6.3 in both human liver mitochondria and microsomes (Fig. 2). The presence of potassium phosphate in the reaction buffer revealed no significant difference in the reaction profile as compared with the use of Tris-buffer. Phosphate buffer, pH 6.3, was used in further experiments. The reaction showed linearity with protein and time (data not shown), and followed Michaelis-Menten kinetics. A higher enzymatic rate of N-hydroxymelagatran reduction was seen in human liver mitochondria as compared with microsomes (Fig. 3A). The apparent KM for the reaction was 0.7 to 1 mM. NADH was a much better electron donor than NADPH (Fig. 3, B and C), and in human liver microsomes, an ∼4-fold higher Vmax and an ∼2-fold lower KM were seen using NADH as compared with NADPH (Fig. 3B). Similar results were observed in human liver mitochondria (Fig. 3C).

Effect of pH on the NADH-dependent reduction of N-hydroxymelagatran to melagatran. Both potassium phosphate buffer and Tris-buffer were used in the reaction analyses. Results displayed are mean values of duplicate samples, with error bars in SM.

Effect of substrate concentration (A), and NADH and NADPH (B and C) on the reduction of N-hydroxymelagatran in human liver microsomes and mitochondria. Results displayed are mean values of duplicate samples, with error bars in SM. The kinetics were visualized using Sigma Plot Kinetics Module for Windows 7.0.
Substrate Specificity. Competition experiments were carried out using benzamidoxime and N-methylhydroxylamine as inhibitors of the reduction of N-hydroxymelagatran (Fig. 4). Benzamidoxime competed with the reduction of N-hydroxymelagatran in both human liver mitochondria and microsomes (Fig. 4A), whereas N-methylhydroxylamine exerted an IC50 of 11.9 ± 1.16 μM for N-hydroxymelagatran reduction in human liver microsomes (Fig. 4B) and an IC50 of 17.3 ± 1.07 μM in liver mitochondria (Fig. 4C). The data indicate that benzamidoxime and N-methylhydroxylamine are reduced by the same enzyme system as N-hydroxymelagatran.
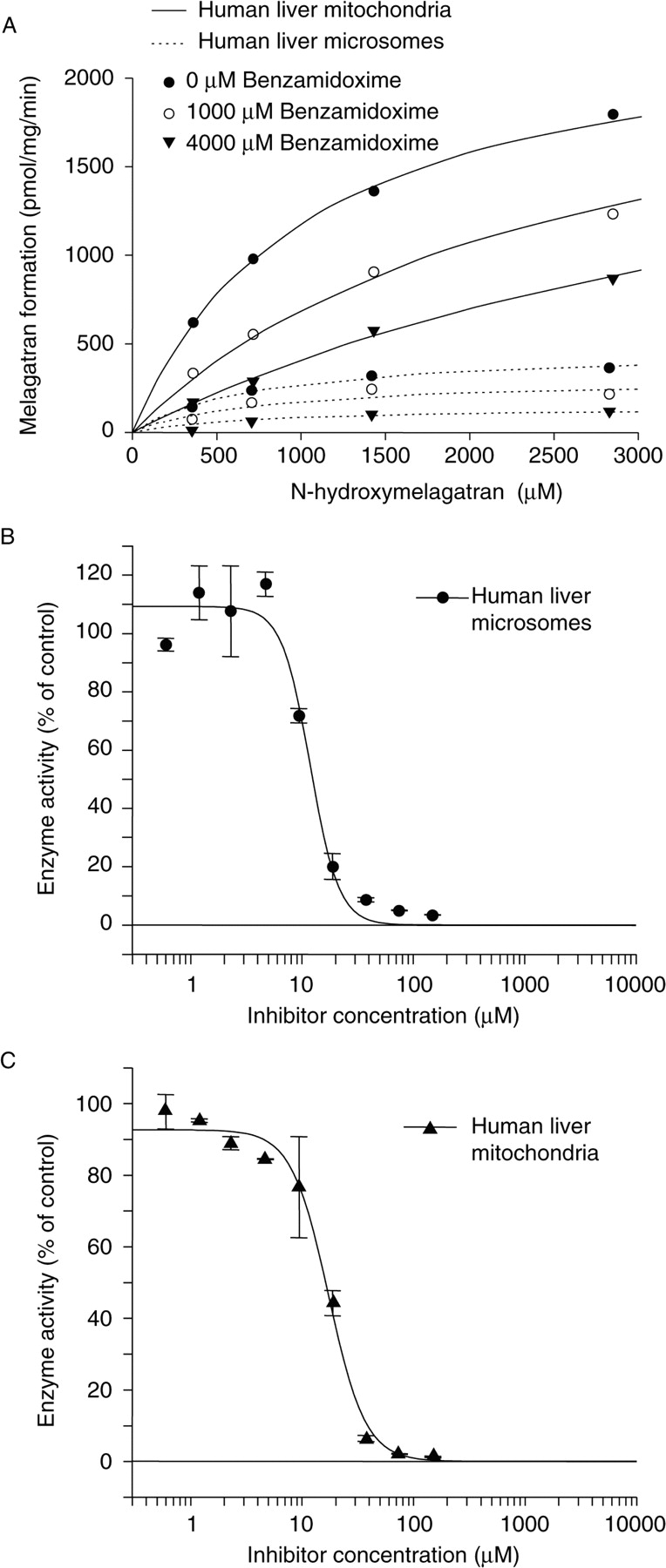
Inhibition of reduction of N-hydroxymelagatran by benzamidoxime (A) and N-methylhydroxylamine (B and C). One hundred percent activity corresponds to 389.5 ± 45.6 pmol/mg/min in human liver microsomes (B) and 1109.4 ± 95.5 pmol/mg/min in human liver mitochondria (C). Results displayed are mean values of duplicate samples, with error bars in SM.
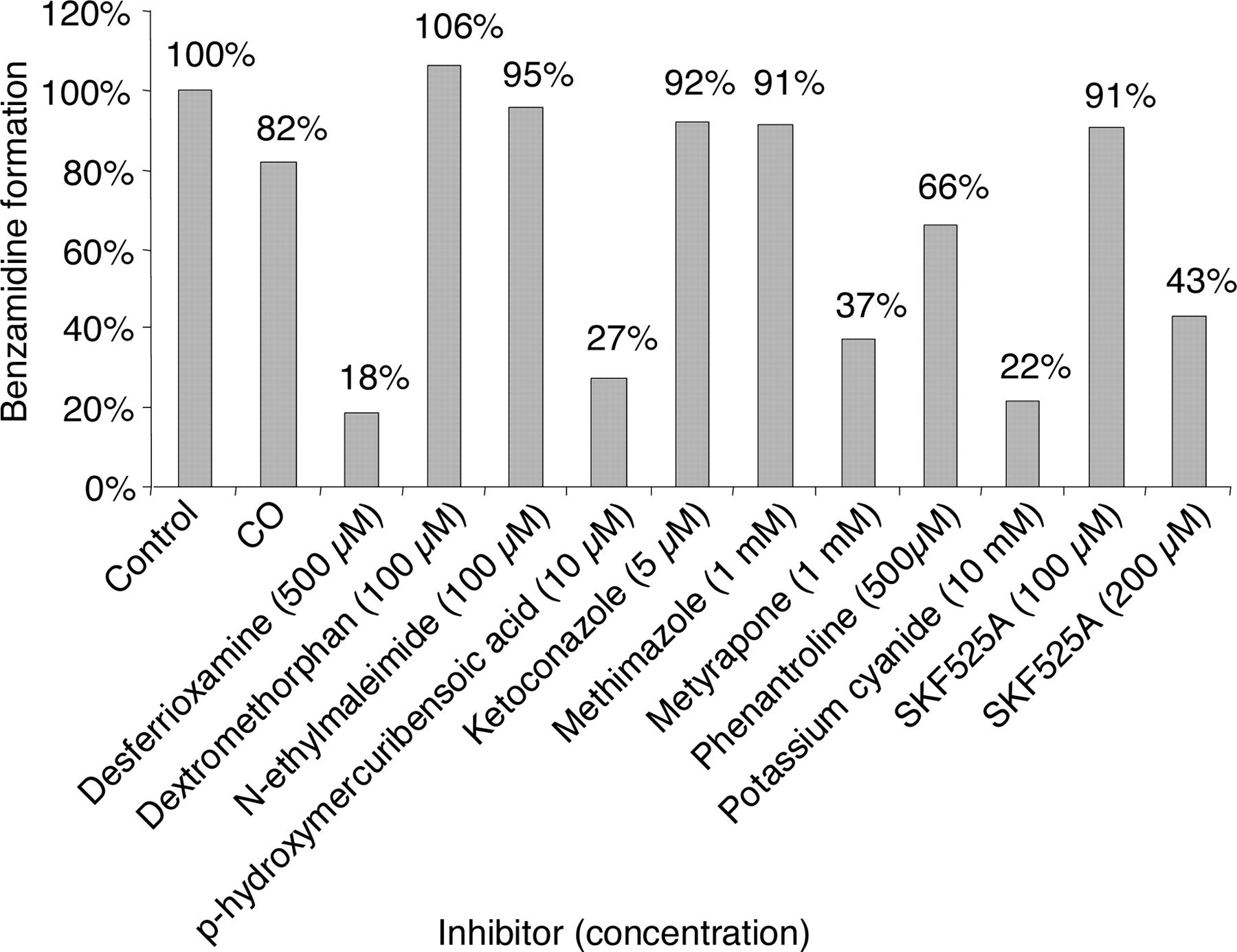
Inhibition Studies. Neither CO treatment nor concomitant incubation with dextromethorphan, ketoconazole, methimazole, N-ethylmaleimide, or lower concentrations of SKF525A inhibited the reduction of benzamidoxime. At low concentration, no inhibition was seen using SKF525A, whereas some inhibition was evident at higher concentrations using metyrapone and SKF525A (Fig. 5). The benzamidoxime reductase was, in contrast, strongly inhibited by desferrioxamine, p-hydroxymercuribenzoic acid, and KCN, indicating that P450 enzymes do not contribute to the reduction of N-hydroxylated amidines.

Effect of various inhibitors on the rate of NADH-dependent benzamidoxime reduction in human liver microsomes. Incubation conditions were as described under Materials and Methods. Four different human livers were used in the inhibition experiments except for CO, and the inhibitors were evaluated in at least two different livers. The results are given as the mean values from two to four different livers used for each inhibitor.
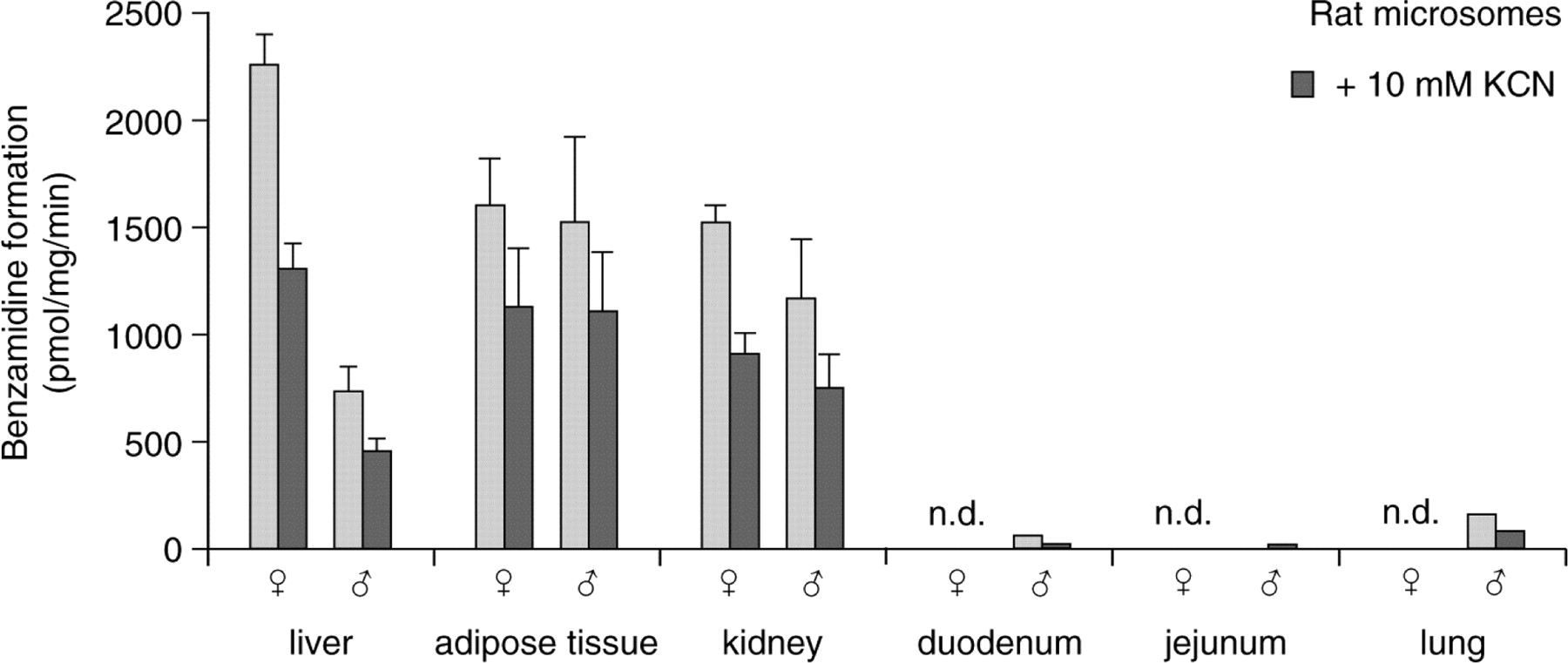
Tissue Distribution and Sex Differences in Rats. The benzamidoxime reductase and N-hydroxymelagatran reductase activities were seen in a variety of different rat tissues (Fig. 6). The highest activity was observed in liver, kidney, and adipose tissue. A sex difference was noted in rat liver, where females (8–9 weeks old) had about a 4-fold higher benzamidoxime reductase activity than males of the same age, whereas no significant sex differences were seen using microsomes from adipose tissue, kidneys, duodenum, jejunum, or lung. In general, 10 mM KCN inhibited the activity seen in the rat microsomal preparations by 40 to 50%.

The rate of benzamidoxime reduction in rat microsomes isolated from different tissues. Both female and male microsomes were analyzed in the absence or in the presence of 10 mM KCN. For tissues with high enzyme expression, the standard deviation is marked, and results are based on three or more individual rat samples incubated more than two times in at least duplicate. Results from low-expression tissues are based on one experiment with two rats in each group. n.d., not determined.
Tissue Distribution in Humans. The benzamidoxime reductase and N-hydroxymelagatran reductase activities were most abundant in microsomes and mitochondria derived from liver. In contrast to the rat, human liver mitochondria had 3- to 4-fold higher specific activities compared with microsomes, using both substrates (Fig. 7, A and B). In commercially available human adipose tissue, the mitochondrial activity was 5 to 10 times less than in liver, and almost absent in the human adipose tissue microsomes. All the subcellular fractions originating from human tissues showed strong, almost 90% inhibition by 10 mM KCN (Fig. 7B).

The rate of N-hydroxymelagatran (A) and benzamidoxime (B) reduction in human microsomes (mic.) and mitochondria (mit.) isolated from different tissues. The enzyme activity was analyzed in the absence or in the presence of 10 mM KCN.
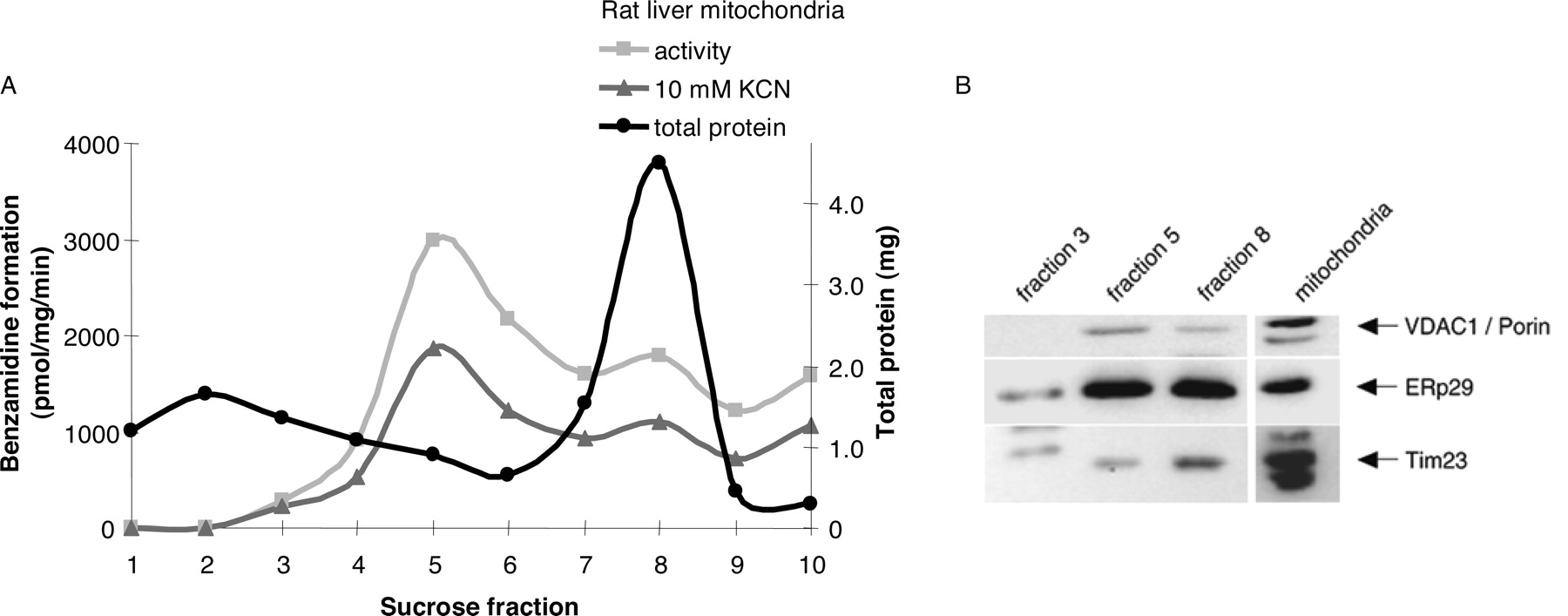
Submitochondrial Localization in Rats. The separation visualized three orange-reddish colored bands, the uppermost just entering the 23.2% (w/v) sucrose fraction, and the other two in the intersections between the 23.2 and 37.4% sucrose fractions, and between the 37.4 and 51.3% sucrose fractions. As shown in Fig. 8A, the fraction corresponding to outer mitochondrial membrane (fraction 5) contained the highest specific activity when compared with fraction 8 residing in the inner membrane. Western blotting analysis using specific antibodies toward microsomal marker proteins, as well as proteins of the inner and outer mitochondrial membranes, confirmed the separation of inner and outer membranes by the sucrose gradient treatment (Fig. 8B). The highest amounts of inner mitochondrial membrane Tim23 were detected in fraction 8, whereas the outer membrane anti-VDAC1/porin was present mainly in fraction 5. These data indicate a preferential localization of the benzamidoxime reductase to the outer mitochondrial membranes.

Fractionation of rat liver mitochondria on a sucrose gradient (A) and Western blotting analyses (B) of the fractions using specific antibodies against inner mitochondrial membrane (anti-Tim23), outer mitochondrial membrane (anti-VDAC1/porin), and microsomal cross-contamination (anti-ERp29). The results shown are based on a single experiment, with incubations made in duplicate with and without KCN inhibition for each fraction. In B, 20 μg of protein sample were loaded per lane.
Protein Fractionation by PEG 6000 and Octyl Sepharose. To purify the reductase, rat (Fig. 9) as well as human (Fig. 10) microsomal and mitochondrial fractions were subjected to stepwise PEG 6000 protein precipitation. In both rat liver microsomes and mitochondria, the highest specific activity was seen in the 15 to 18% (w/v) PEG fraction. Fractions from rat liver mitochondria had an ∼2.5-fold higher specific activity level than did rat liver microsomes. Immunostaining of the respective mitochondrial PEG fractions with specific antibodies exposed a massive accumulation of mitochondria outer membrane protein VDAC1/porin, with its peak in fractions containing 9 to 12% and 12 to 15% PEG (Fig. 9E). In parallel, minor amounts of inner mitochondria membrane marker Tim23 were detected in these fractions, whereas the highest amounts of Tim23 were found in 0 to 12% PEG fractions.

PEG fractionation (A and B) and subsequent Octyl Sepharose chromatography of the benzamidoxime reductase activity using rat liver microsomes (A and C) and mitochondria (B and D). The PEG fractionation from rat liver mitochondria and microsomes was also monitored using Western blotting of marker proteins (E), each lane containing 20 μg of protein. Results displayed are from one representative experiment out of several with similar results, with incubations carried out in duplicate. Light bars in the figure indicate enzyme activity; dark bars indicate KCN-inhibited enzyme activity (pmol/mg/min).

PEG fractionation (A and B) and subsequent Octyl Sepharose chromatography of the benzamidoxime reductase activity using human liver microsomes (C). The PEG fractionation was also monitored using Western blotting of marker proteins (D), each lane containing 20 μg of protein. The activity using human liver mitochondria was too low for detection after the Octyl Sepharose column. Results displayed are from one representative experiment out of several with similar results, with incubations carried out in duplicate. Light bars in the figure indicate enzyme activity; dark bars indicate KCN-inhibited enzyme activity (pmol/mg/min).
The PEG-precipitated protein fractions with the highest specific activity (15–18% PEG) were further chromatographed on an Octyl Sepharose column (Fig. 9, C and D). The benzamidoxime reductase activity was eluted in slightly turbid noncolored fractions of the void volume and was thereby separated from the bulk of proteins retarded on the column.
For microsomes and mitochondria derived from human liver, the highest reductase activity was seen in the 12 to 15% PEG fractions (Fig. 10, A and B). Similarly, as with the rat fractions, Tim23 accumulated in fractions with low PEG concentrations (Fig. 10D). Human liver mitochondrial 9 to 12% and 12 to 15% PEG fractions had a high specific reductase activity (Fig. 10B) and high amounts of outer membrane protein VDAC1/porin (Fig. 10D).
The 12 to 15% PEG human microsomal fraction showed the same fractionation pattern on Octyl Sepharose as the rat liver microsomal proteins, indicating that similar proteins had been monitored in both species.
The partial purification process was monitored by the reduction of total protein content rather than following specific activity, since every solubilization led to a reduction in enzymatic activity. All attempts to further purify the enzyme using a variety of nonionic and ionic detergents, and ion exchange columns were unsuccessful in that no enzyme activity could be recovered. This result indicates an extreme instability of the enzyme when released from the protein complex.
Discussion
In this study, we have characterized and partially purified the enzyme system active in the reduction of N-hydroxymelagatran and benzamidoxime from both rat and human liver microsomes and mitochondria. Its instability prevented us from achieving purification of the system to an appreciable purity. In vitro incubations performed with concomitant addition of N-hydroxymelagatran, N-methylhydroxylamine, and benzamidoxime showed an inhibition of the N-reduction in both human liver microsomes and mitochondria in a manner indicating the interaction of all compounds with the same enzyme system. The enzyme system has a unique pH optimum of 6.3, also seen for the NADH-dependent reduction of sulfamethoxazole hydroxylamine in dog and human liver microsomes (Trepanier and Miller, 2000) and the NADH-dependent reduction of N-hydroxylamine (Kadlubar et al., 1973).
From our inhibition studies (Fig. 5), we conclude that the reductase system does not have cytochrome P450 as an essential component due to the apparent lack of inhibition detected using several different cytochrome P450 inhibitors. SKF525A, interfering with the P450 substrate binding, often causing type I spectral change, showed no inhibition at lower concentrations (100 μM), and only demonstrated partial inhibition in the higher concentration range (200 μM). CO and metyrapone, two inhibitors binding to the heme iron of the cytochrome P450, did not inhibit, or slightly inhibited the activity, respectively. Neither did the results using ketoconazole nor the results using dextromethorphan indicate that the reduction should involve cytochrome P450. Furthermore, we obtained a very high specific enzyme activity in adipose tissue from rats. Indeed the specific activity of the reductase reaction here was the highest among all male rat tissues examined (Fig. 6), in a tissue where essentially no cytochrome P450 is present. All these data strongly indicate the absence of any participation of cytochrome P450 in the reduction of benzamidoxime and N-hydroxymelagatran. The results are not compatible with data previously presented, in which the participation of a CYP2D enzyme in an electron transport chain also including NADH-cytochrome b5 reductase and cytochrome b5, was suggested (Clement el al., 1997; Clement and Lopian, 2003). Furthermore, Lin et al. (1996) failed to obtain a correlation between the rate of hydroxylamine reduction (using 10-N-(8-(hydroxylamino)octyl)-2-(trifluoromethyl)phenothiazine) and the presence of CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 in human liver microsomes. In our own experiments, we failed to reconstitute the reductase activity using recombinant CYP1A1, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, or CYP3A5 (T. B. Andersson, unpublished observations). Thus, further work is needed to elucidate the discrepancy between the results presented.
It is also unlikely that the reductase system belongs to the group of flavine monooxygenases (FMOs), since no loss of activity was seen in the presence of 1 mM methimazole (Fig. 5), a high affinity substrate for the FMOs (Poulsen et al., 1974). Nor was the enzyme stabilized by the presence of cofactor (NADH) when pretreated by moderate heat, as the heat-sensitive FMO would (Ziegler and Pettit, 1966), when assayed after the protocol of Grothusen et al. (1996) (results not shown), and no correlation between FMO and the hydroxylamine reductase activity has been seen in human liver microsomes (Lin et al., 1996).
Incubations with different thiol blocking agents showed no inhibition by N-ethylmaleimide, but a potent suppression of activity was seen using 10 μM p-hydroxymercuribenzoate, considered to be a relatively specific inhibitor at these concentrations of NADH-cytochrome b5 reductase (Shimada et al., 1998). This supports the original suggestion that the reduction of N-hydroxylamine requires NADH-cytochrome b5 reductase (Kadlubar and Ziegler, 1974).
We obtained strong indications for the participation of nonheme iron in the reduction, since the activity was readily inhibited by the iron chelator desferrioxamine, as well as by KCN. A smaller extent of cyanide inhibition and, in some cases, even activation, was seen using preparations accumulated with inner mitochondrial membrane, most probably due to inhibition of cytochrome oxidase and subsequent uncoupling of electrons from the membranes reaching components that were able to reduce the substrates in a more unspecific fashion.
We observed an unexpected sex difference in rats, in that the rate of NADH-dependent reduction of benzamidoxime was 4-fold higher in liver microsomes from female as compared with male microsomes. By contrast, no sex differences were observed in microsomes from rat adipose tissue or kidneys. The specific activity toward benzamidoxime was highest in liver microsomes from female rats of all the membranes investigated (Fig. 6), although microsomes from adipose tissue had the highest specific activity in male rats. The existence of hepatic gender-predominant expression patterns in rats is well known for some enzymes belonging to the cytochrome P450 family, whereas similar sex differences are not present in human liver (Shapiro et al., 1995). Comparatively much lower activity was seen using commercial preparations of human adipose tissue when incubated with N-hydroxymelagatran (Fig. 7). The finding of the enzyme system in adipose tissue was unexpected and very interesting since, to our knowledge, no drug metabolism has previously been shown to be located in this tissue to such a high specific activity. This finding indicates that the enzyme system might be involved in the lipid metabolism.
We provide evidence for a localization of the rat reductase system to the outer mitochondrial membranes as revealed by sucrose gradient fractionation of rat liver mitochondrial membranes (Fig. 8). After PEG fractionation, the mitochondrial preparations contained about 2.4-fold higher specific activity as compared with microsomal preparations, whereas in human preparations, the activities were similar (Figs. 9 and 10). Less cyanide inhibition in the fractionation of human mitochondria was obtained in early PEG fractions, in which inner membrane proteins were present according to Western blotting analysis using Tim23 antibodies. This result might indicate that cytochrome oxidase contributes to the activity in the PEG 0 to 6% and 6 to 9% fractions (Fig. 10, B and D). In general, the fractions from human microsomes and mitochondria were much more sensitive to cyanide.
Octyl Sepharose column chromatography as a purification step after the PEG fractionation was partially successful. The enzyme activity eluted in white turbid fractions in the void volume separated well from the bulk of proteins in the PEG fractions. The proteins in the void fractions existed as aggregates, as revealed by microscopic examination and by chromatography on Sephacryl (data not shown). Upon solubilization of these aggregates, the activity was lost. This represents a major obstacle for future attempts to purify the enzyme further.
In conclusion, we have partially purified the system responsible for the reduction of amidoximes and shown its tissue and intracellular distribution. This system appears to be similar to that active in the reduction of N-hydroxylamines. This system, which is cyanide-sensitive and NADH-dependent, is active in the prodrug activation of ximelagatran (Exanta) and represents a very atypical example of a non-P450-dependent drug-metabolizing system because of its high specific activity in adipose tissue. The determination of its exact nature requires further investigations.
Acknowledgments
We are grateful to Dr. Souren Mkrtchian, Division of Molecular Toxicology, Institute of Environmental Medicine, Karolinska Institutet, for providing the polyclonal anti-ERp29 antibody. We also thank the biomedicine students Christin Bexelius and Maria Pernemalm for initial work with adipose tissue. The anti-Tim23 antibody was a kind gift from M. F. Bauer and S. Hofmann at University of Munich, Germany.
Footnotes
-
Funding for this study was provided by AstraZeneca AB, by the Swedish Research Council, and by Magn. Bergvalls Stiftelse. Parts of this work have been presented at the 7th International ISSX Meeting, August 29-September 2, 2004, in Vancouver, Canada.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002972.
-
ABBREVIATIONS: CO, carbon monoxide; FMO, flavine monooxygenase; HPLC, high-pressure liquid chromatography; LC-MS, liquid chromatography-mass spectrometry; PEG, polyethylene glycol; KCN, potassium cyanide; PMSF, phenylmethylsulfonyl fluoride; psig, pounds per square inch gauge; P450, cytochrome P450; SKF525A, b-diethylaminoethyldiphenylpropylacetate hydrochloride.
- Received November 11, 2004.
- Accepted January 4, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}