


Abstract
Methylcarbamate benzimidazoles [albendazole (ABZ), fenbendazole (FBZ), and their respective sulfoxide derivatives, albendazole sulfoxide (ABZSO) and oxfendazole (OXF)] are therapeutically important anthelmintic agents with low bioavailability. We studied their in vitro interaction with the apical ATP-binding cassette (ABC) drug efflux transporters, breast cancer resistance protein (BCRP/ABCG2), P-glycoprotein (ABCB1), and MRP2 (ABCC2) using MDCKII cells transduced with human MDR1, MRP2, and BCRP, and murine Bcrp1 cDNAs. These ABC drug efflux transporters extrude a wide range of xenotoxins from cells in intestine, liver, and other organs, thus affecting the bioavailability of many compounds. In transport experiments, ABZSO and OXF were transported efficiently by murine Bcrp1 and moderately by human BCRP, but not by MDR1 or MRP2. ABZ and FBZ were not found to be Bcrp1, MRP2, or P-glycoprotein substrates in vitro. OXF was found to be a good BCRP/Bcrp1 inhibitor, with somewhat higher potency in the MDCKII-BCRP cell line. The latter results were confirmed by flow cytometry experiments demonstrating inhibition by OXF of murine Bcrp1- and human BCRP-mediated mitoxantrone transport. Further studies of interactions between OXF and known BCRP/Bcrp1 substrates will be of interest. The use of efficacious BCRP/Bcrp1 inhibitors might increase the extent and duration of systemic exposure to ABZSO and OXF, with possible therapeutically beneficial effects in extra-intestinal infections.
Albendazole (ABZ), fenbendazole (FBZ), and their sulfoxide derivatives (albendazole sulfoxide/ricobendazole, ABZSO; and fenbendazole-sulfoxide/oxfendazole, OXF) are methylcarbamate benzimidazoles with a broad-spectrum anthelmintic activity, widely used in human and veterinary medicine. They are used against several systemic parasitoses including nematodoses, hydatidosis, taeniasis, and others (Campbell, 1990). They are also active against various protozoa (Katiyar et al., 1994) and Gram-positive bacteria (Elnima et al., 1981). ABZ is used to treat microsporidial and cryptosporidial infections, which can cause lethal diarrhea in patients treated with immunosuppressive drugs or those infected with HIV (Zulu et al., 2002; Didier et al., 2004). In addition, ABZ has shown antitumor activity (Morris et al., 2001; Pourgholami et al., 2004). Both sulfoxide derivatives (ABZSO and OXF) are the main active metabolites found in plasma after oral administration of the parent compounds, ABZ and FBZ (Lanusse and Prichard, 1993), but they are also available as commercial formulations themselves.
Especially FBZ, but also ABZ, and their sulfoxides are poorly absorbed from the gastrointestinal tract (Lanusse and Prichard, 1993; Lanusse et al., 1995; Capece et al., 2000). For treatment of intestinal luminal parasites, this is ideal: intestinal concentration of the drug remains high, and there is little risk of systemic toxicity. However, for systemic treatments elsewhere in the body, substantial (systemic) uptake of the drugs is needed, and low benzimidazole bioavailability is a disadvantage. ABZ and FBZ are known to cross plasma membranes by passive diffusion due to their lipophilicity (Mottier et al., 2003), but the existence of additional (uptake) transport mechanisms cannot be excluded.
Low water solubility of benzimidazoles has been considered a limiting factor in their low bioavailability (Capece et al., 2000), but for many drugs, it has been shown that their oral availability is also reduced by ATP-binding cassette (ABC) drug efflux transporters present in the apical membrane of intestinal epithelia (among other epithelia) (Zhu, 1999; Jonker et al., 2000). Significant direct secretion of ABZSO into the intestinal lumen has been demonstrated (Redondo et al., 1999; Merino et al., 2003). The understanding of the mechanism involved in the low availability of these compounds might lead to the design of new strategies to modify this pharmacokinetic property when desired and, thus, enhance therapeutic efficacy in systemic treatments.
In this paper, we studied the in vitro transport of some therapeutically relevant methylcarbamate benzimidazoles by apical ABC transporters [breast cancer resistance protein (BCRP/ABCG2), P-glycoprotein (P-gp/ABCB1), MRP2/ABCC2] using MDCKII cells transduced with human MDR1, MRP2, and BCRP, and murine Bcrp1 cDNAs.
Materials and Methods
Chemicals. ABZ and MBZ were obtained from Sigma-Aldrich (St. Louis, MO). ABZSO was kindly supplied by GlaxoSmithKline (Madrid, Spain). FBZ and OXF were supplied by Rhône Mérieux (Lyon, France). PhIP and [14C]PhIP (10 Ci/mol) were obtained from Toronto Research Chemicals, Inc. (Toronto, ON, Canada). [3H]Inulin was obtained from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). Ko143 was described previously as a Bcrp1 and BCRP inhibitor (Allen et al., 2002). All other compounds used were reagent grade.
Transport Assays. Generation, culturing, and characterization of human MDR1-, MRP2-, and BCRP-, and murine Bcrp1-transduced MDCKII subclones were previously described (Evers et al., 1998; Jonker et al., 2000; Pavek et al., 2005). Transepithelial transport assays were carried out as described (Huisman et al., 2001). Appropriate positive controls were used for each transfected cell line to verify optimal transport performance. Inhibition of BCRP/Bcrp1-mediated transport was determined after the addition of 325 μM OXF or 5 μM Ko143 to both the apical and basal compartments, using [14C]PhIP as BCRP/Bcrp1 substrate (van Herwaarden et al., 2003; Pavek et al., 2005). At the end of the experiment, filters were excised and measured for radioactivity.
Accumulation Assays. In vitro accumulation assays were carried out as described previously (Pavek et al., 2005). Mitoxantrone (5 μM) was used as fluorescent substrate and OXF was used as inhibitor at different concentrations. Relative cellular accumulation of mitoxantrone was determined by flow cytometry using a FACSCalibur cytometer in at least 5000 cells from histogram plots using the median of fluorescence (MF). Inhibitory potencies of compounds were calculated as described (Pavek et al., 2005) in MDCKII-BCRP or MDCKII-Bcrp1 cells according to the following equation: inhibitory potency = (MF with OXF–MF without inhibitor)/(MF with Ko143–MF without inhibitor) × 100%.
HPLC Analysis. HPLC analysis of the samples from the transport assays was based on a published method (Virkel et al., 2004) with minor modifications. Culture medium (100 μl) was directly injected into the HPLC system. Separation was performed on a reversed-phase column (Nucleosil 120 C18, 10 μm; 250 mm × 4 mm). The composition of the mobile phase was acetonitrile/ammonium acetate (0.025 M, pH 6.6) at a proportion of 67:33 for ABZSO and OXF, 39:61 for ABZ, and 50:50 for FBZ. The flow rate was set to 1 ml/min for ABZSO and OXF, 0.8 ml/min for ABZ, and 1.5 ml/min for FBZ. UV absorbance was measured at 292 nm.
Results and Discussion
To investigate whether methylcarbamate benzimidazoles are efficiently transported by apical ABC transporters, we determined the involvement of P-gp, MRP2, and Bcrp1 in ABZ, FBZ, ABZSO, and OXF transport in vitro, using the polarized canine kidney cell line MDCKII and its subclones transduced with human MDR1 and MRP2, and murine Bcrp1 cDNAs. In addition, human BCRP-transduced MDCKII subclones were tested for the possible role of human BCRP in the in vitro transport of ABZSO and OXF. The parental and transduced cell lines were grown to confluent polarized monolayers on porous membrane filters, and vectorial transport of the tested compound (10 μM) across the monolayers was determined.
For ABZSO and OXF, apically directed translocation was highly increased and basolaterally directed translocation drastically decreased (Fig. 1, A, B, E, and F) in the Bcrp1-transduced compared with the MDCKII parental cell line. When the selective Bcrp1 inhibitor Ko143 was used (Allen et al., 2002), this Bcrp1-mediated transport was completely inhibited (Fig. 1, C and G), resulting in a vectorial translocation pattern equal to that of the MDCKII parental cell line. When the human BCRP-transduced cell line was used, the difference in the directional transport was lower than in the case of the murine Bcrp1-transduced cell line (Fig. 1, D and H) for both ABZSO and OXF. In contrast, in the MDR1- and MRP2-transduced MDCKII cell lines, the vectorial translocation was similar to that in the MDCKII parental cell line (Fig. 2, A and B). No significant vectorial transport was observed for ABZ and FBZ in MDCKII and its subclones transduced with murine Bcrp1 and human MDR1 and MRP2 cDNAs (Fig. 2, C and D).
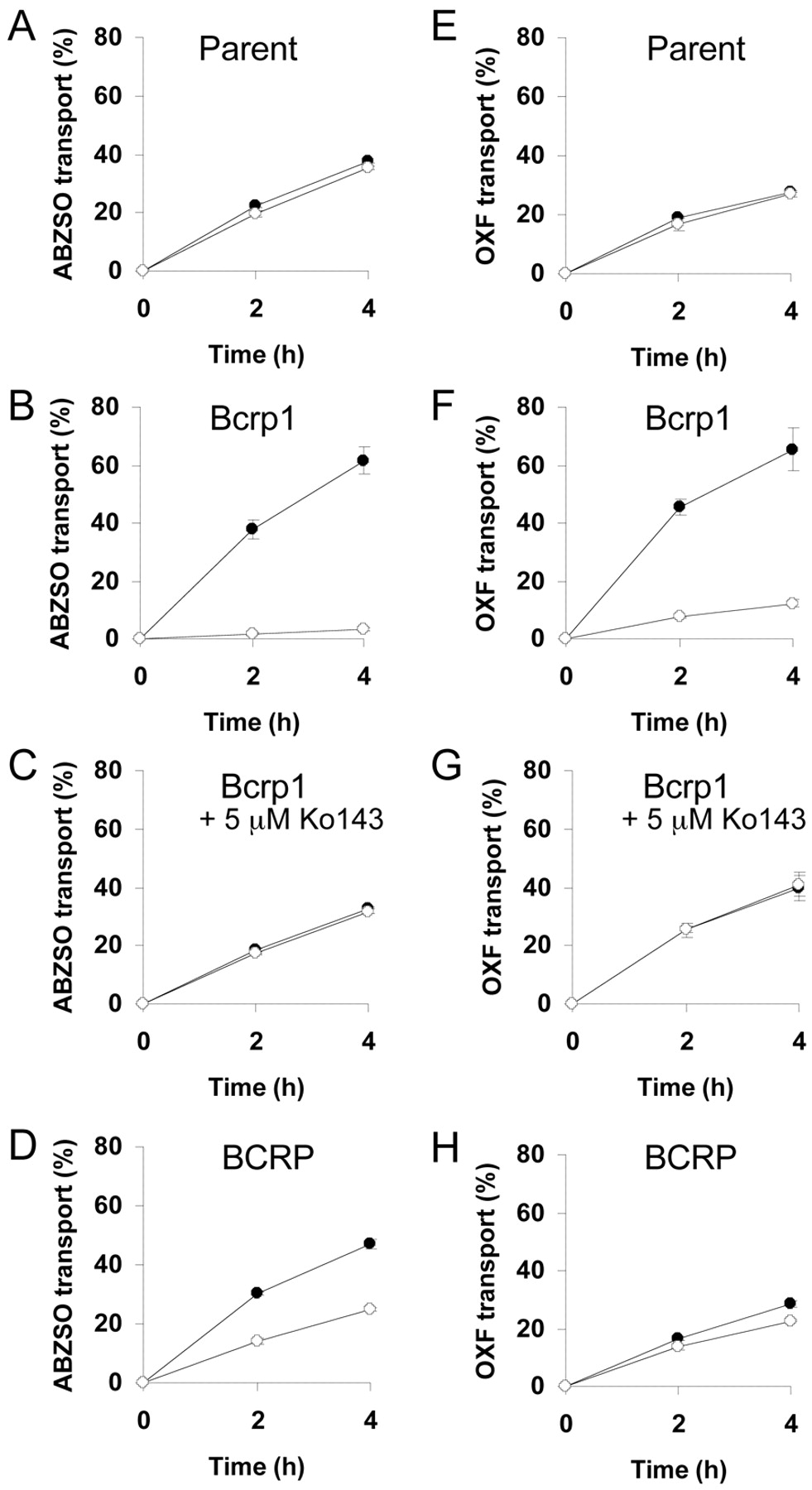
Transepithelial transport of ABZSO (10 μM, left panel) and OXF (10 μM, right panel) in MDCKII (parent) (A and E), MDCKII-Bcrp1 (B and C, F and G), and MDCKII-BCRP (D and H) monolayers. The experiment was started with the addition of ABZSO or OXF to one compartment (basolateral or apical). After 2 and 4 h, the percentage of drug appearing in the opposite compartment was measured by HPLC and plotted. BCRP inhibitor Ko143 (C and G) was present as indicated. Results are the means; error bars indicate the standard deviations (n = 3). Closed circles, translocation from the basolateral to the apical compartment; open circles, translocation from the apical to the basolateral compartment. The apparent permeability coefficient (Papp) corresponding to 10% of transport in 1 h was 12.3 · 10–6 cm/s.
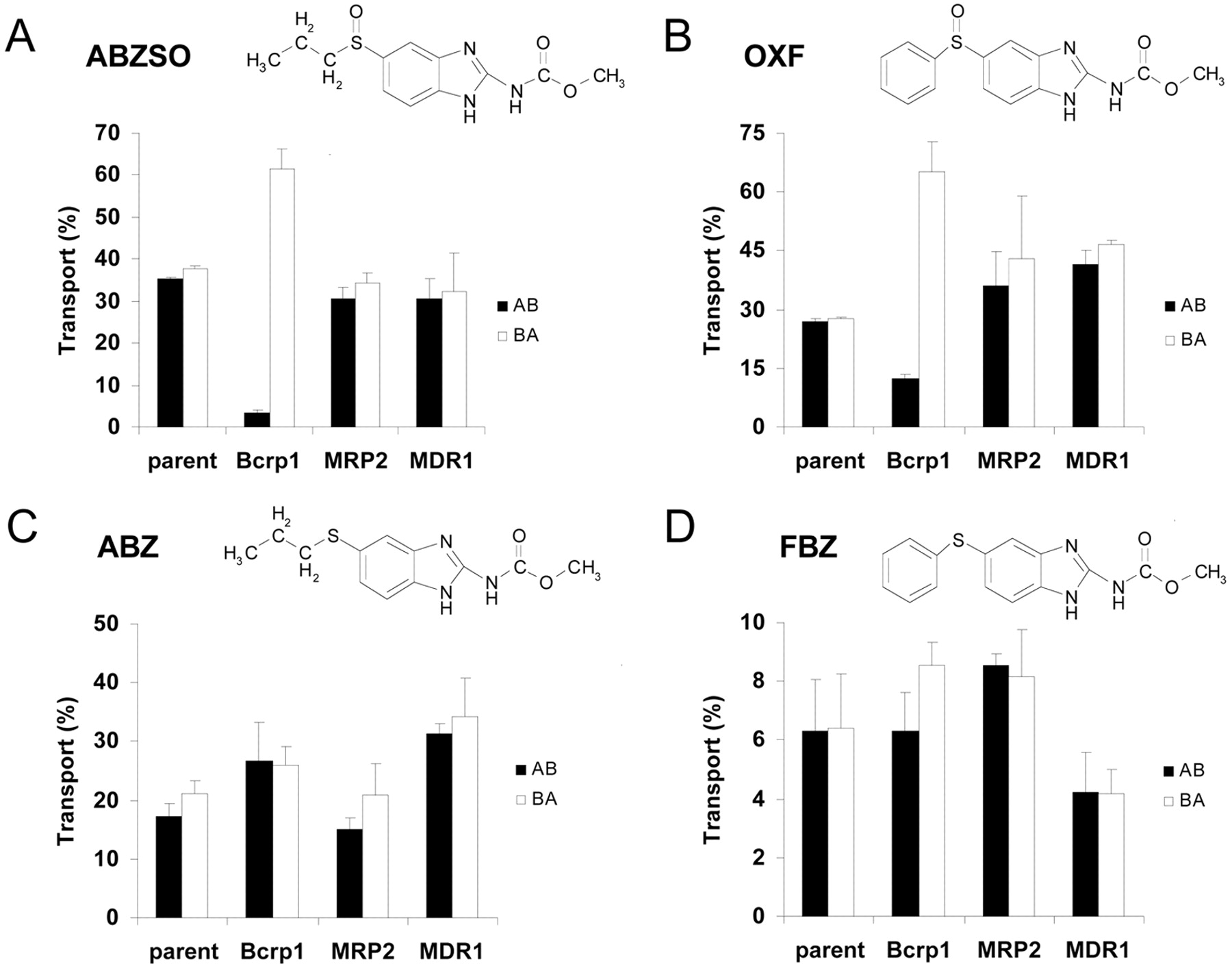
Transepithelial transport of ABZSO (A), OXF (B), ABZ (C), and FBZ (D) in MDCKII (parent), MDCKII-Bcrp1, MDCKII-MRP2, MDCKII-MDR1, and monolayers. The experiment was started with the addition of the tested compound (10 μM) to one compartment (basolateral or apical). After 4 h, the percentage of drug appearing in the opposite compartment was measured by HPLC and plotted. Results are the means; error bars indicate the standard deviations (n = 3). BA, translocation from the basolateral to the apical compartment; AB, translocation from the apical to the basolateral compartment. The Papp corresponding to 10% of transport in 1 h was 12.3 · 10–6 cm/s. Chemical structures of the compounds and some data (A and B, MDCKII-Bcrp1) from Fig. 1 are included for comparison.
Together, these results show highly efficient transport of ABZSO and OXF by murine Bcrp1, moderate transport by human BCRP, and no transport by MDR1 or MRP2. ABZ and FBZ are not Bcrp1, MRP2, or P-gp substrates in vitro. This is in agreement with the lack of P-gp involvement in ABZ transepithelial transport using LLC-PK1 cells transfected with MDR1 cDNA obtained by Merino et al. (2002).
It is interesting to note that the simple sulfoxidation of ABZ and FBZ converts non-Bcrp1 substrates into well transported Bcrp1 substrates (Fig. 2, A to D), even though for FBZ, the overall transcellular permeability (percentage of transport) in the parent cells was also increased by this modification (Fig. 2, B and D).
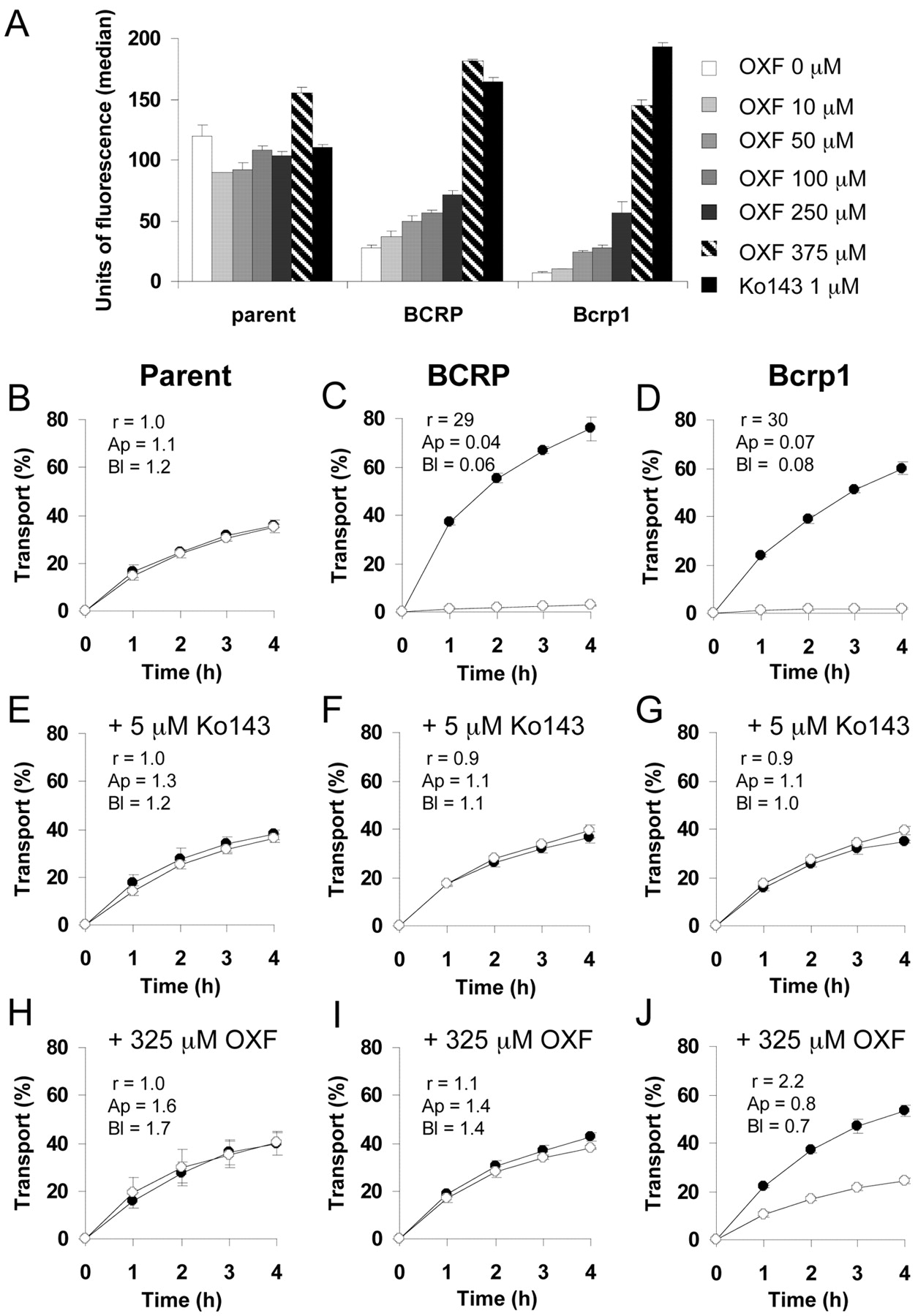
To further characterize the interactions of the methylcarbamate benzimidazoles with Bcrp1/BCRP, the ability of these compounds to modulate mitoxantrone accumulation in murine Bcrp1- and BCRP-expressing cell lines was tested in flow cytometry experiments. BCRP inhibition increases the accumulation of mitoxantrone in Bcrp1- and BCRP-transduced cells and thus increases the MF. OXF had the strongest inhibitory potency in both cell lines (75.2 ± 2.5% in MDCKII-Bcrp1 and 112.7 ± 1.2% in MDCKII-BCRP at a concentration of 375 μM; Fig. 3A), whereas ABZSO only reached an inhibitory potency of 6.9 ± 0.4% in MDCKII-Bcrp1 and 28.9 ± 2.6% in MDCKII-BCRP at a concentration of 375 μM (not shown). ABZ and FBZ had an inhibitory potency of less than 15% at 50 μM (limit of solubility) in both cell lines (not shown).

OXF is a BCRP/Bcrp1 inhibitor in vitro. A, effect of different concentrations of OXF on accumulation of mitoxantrone in parent MDCKII cells and in their BCRP- and Bcrp1-transduced derivatives, preincubated with or without Ko143 or OXF at the indicated concentrations. Results are expressed as means; the error bars indicate the standard deviations from at least three experiments. B to J, transepithelial transport of [14C]PhIP (2 μM) across MDCKII-parent (B, E, and H), MDCKII-BCRP (C, F, and I), and MDCKII-Bcrp1 (D, G, and H) monolayers. The experiment was started with the application of [14C]PhIP to one compartment (basal or apical). The percentage of radioactivity appearing in the opposite compartment was measured and plotted. The BCRP inhibitor Ko143 (E–G) or OXF (H–J) was added in both compartments. Results are presented as the means; the error bars indicate the standard deviations from at least three experiments. Closed circles, translocation from the basolateral to the apical compartment; open circles, translocation from the apical to the basolateral compartment. r, transport ratio (ratio of apically directed translocation divided by the basolaterally directed translocation by the end of the experiment); Ap and Bl, percentage of total radioactivity applied to the apical (Ap) or basolateral (Bl) compartment that was retrieved from the cell layer. The Papp corresponding to 10% of transport in 1 h was 12.3 · 10–6 cm/s.
Since OXF was the most potent inhibitor of Bcrp1, its inhibitory potential on transepithelial transport of the dietary carcinogen PhIP was tested. PhIP is an excellent BCRP/Bcrp1 substrate (van Herwaarden et al., 2003; Pavek et al., 2005). OXF (at 325 μM) completely reversed transepithelial transport of PhIP across MDCKII-BCRP cells, like the potent Bcrp1/BCRP inhibitor Ko143 (at 5 μM), and without significant effect on the parent cells (Fig. 3, B, C, E, F, H, and I). The radioactivity associated with the MDCKII-BCRP cell layer was also 28-fold increased by OXF treatment, in accordance with inhibition of BCRP-mediated efflux (Fig. 3, B, C, H, and I). In the MDCKII-Bcrp1 cell line, OXF markedly reduced transport of PhIP across MDCKII-Bcrp1 cells (reducing the transport ratio from 29 to 2.2) (Fig. 3, D, G, and J). The radioactivity associated with the MDCKII-Bcrp1 cell layer was also 10-fold increased by OXF treatment (Fig. 3, D, G, and J). No signs of compromised condition of the monolayer were observed when OXF was added at 325 μM, since paracellular flux of [3H]inulin to the opposite compartment remained <1.5% of the total radioactivity/h.
These results show that OXF is a BCRP/Bcrp1 inhibitor in vitro, with apparently higher potency in the MDCKII-BCRP cell line compared with MDCKII-Bcrp1. The differences in OXF inhibitory potency and in ABZSO and OXF efficiency of transport between MDCKII-Bcrp1 and MDCKII-BCRP cell lines could be due to differences in the affinity/selectivity for substrates/inhibitors between BCRP and Bcrp1, as has been hypothesized (Mizuno et al., 2004). However, note that mitoxantrone accumulation (without inhibitor) is 3.7-fold higher in the MDCKII-BCRP cell line compared with MDCKII-Bcrp1 (Fig. 3A); thus, we cannot exclude a reduced expression level of the transporter in MDCKII-BCRP cells compared with MDCKII-Bcrp1.
The duration of exposure of the parasite to the benzimidazole drug is a key determinant of efficacy. Consequently, strategies have been developed both experimentally and commercially to extend this exposure period through the development of sustained delivery systems and by modifying the metabolism of the drug in the host species (McKellar and Jackson, 2004). In this study, we demonstrated that ABZSO and OXF are both substrates of BCRP and Bcrp1 in vitro. It may thus be that the clinical use of efficacious BCRP/Bcrp1 inhibitors might improve their oral bioavailability and, hence, also their systemic target exposure, by reducing their intestinal elimination and their hepatobiliary secretion, since Bcrp1 is also expressed in the biliary canalicular membrane of hepatocytes and mediates the hepatobiliary excretion of its substrates (Jonker et al., 2000; van Herwaarden et al., 2003). It will therefore be of interest to further study the possible in vivo effect of BCRP in the pharmacokinetics of these benzimidazole drugs and potential drug interactions with these methylcarbamate benzimidazoles and known BCRP/Bcrp1 substrates in therapeutic-target species (humans, livestock).
Acknowledgments
We thank Antonius E. van Herwaarden for critical reading of the manuscript and Dr. Maarten Huisman from the Academic Medical Center (Amsterdam) for technical assistance.
Footnotes
-
This work was supported by Grant NKI 2000-2271 of the Dutch Cancer Society.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.003319.
-
ABBREVIATIONS: ABZ, albendazole; ABC, ATP-binding cassette; ABZSO, albendazole sulfoxide; FBZ, fenbendazole; HPLC, high performance liquid chromatography; Ko143, 3-(6-isobutyl-9-methoxy-1,4-dioxo-1,2,3,4,6,7,12,12a-octahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indol-3-yl)-propionic acid tert-butyl ester; MF, median of fluorescence; OXF, oxfendazole; Papp, apparent permeability coefficient; PhIP, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine; BCRP, breast cancer resistance protein; P-gp, P-glycoprotein.
- Received December 15, 2004.
- Accepted February 4, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}