


Abstract
Acyl glucuronidation is an important metabolic pathway for fluoroquinolone antibiotics. However, it is unclear which human UDP-glucuronosyltransferase (UGT) enzymes are involved in the glucuronidation of the fluoroquinolones. The in vitro formation of levofloxacin (LVFX), grepafloxacin (GPFX), moxifloxacin (MFLX), and sitafloxacin (STFX) glucuronides was investigated in human liver microsomes and cDNA-expressed recombinant human UGT enzymes. The apparent Km values for human liver microsomes ranged from 1.9 to 10.0 mM, and the intrinsic clearance values (calculated as Vmax/Km) had a rank order of MFLX > GPFX > STFX >> LVFX. In a bank of human liver microsomes (n = 14), the glucuronidation activities of LVFX, MFLX, and STFX correlated highly with UGT1A1-selective β-estradiol 3-glucuronidation activity, whereas the glucuronidation activity of GPFX correlated highly with UGT1A9-selective propofol glucuronidation activity. Among 12 recombinant UGT enzymes, UGT1A1, 1A3, 1A7, and 1A9 catalyzed the glucuronidation of these fluoroquinolones. Results of enzyme kinetics studies using the recombinant UGT enzymes indicated that UGT1A1 most efficiently glucuronidates MFLX, and UGT1A9 most efficiently glucuronidates GPFX. In addition, the glucuronidation activities of MFLX and STFX in human liver microsomes were potently inhibited by bilirubin with IC50 values of 4.9 μM and 4.7 μM, respectively; in contrast, the glucuronidation activity of GPFX was inhibited by mefenamic acid with an IC50 value of 9.8 μM. These results demonstrate that UGT1A1, 1A3, and 1A9 enzymes are involved in the glucuronidation of LVFX, GPFX, MFLX, and STFX in human liver microsomes, and that MFLX and STFX are predominantly glucuronidated by UGT1A1, whereas GPFX is mainly glucuronidated by UGT1A9.
Fluoroquinolone antibiotics, first discovered in the 1960s, are widely used for the treatment of a number of systemic infections; in particular, urinary tract infections (Hooper, 1998). They have excellent in vitro activities against both Gram-positive and Gram-negative organisms and anaerobes (Appelbaum and Hunter, 2000). Newer fluoroquinolones, developed during the 1990s, exhibit enhanced activity against common respiratory pathogens including some drug-resistant strains of Streptococcus pneumoniae (Andersson and MacGowan, 2003). In addition, they show good in vitro activity against atypical pathogens, such as Chlamydia pneumoniae, Mycoplasma pneumoniae, and Legionella pneumophila (Emmerson and Jones, 2003). Most fluoroquinolones have excellent pharmacokinetics, as characterized by their high serum levels, good bioavailability, and extensive distribution into many tissues and body fluids except for cerebrospinal fluid (Dudley, 2003). In humans, many fluoroquinolones are eliminated by glomerular filtration and active tubular secretion in the kidneys; however, hepatic metabolism and biliary excretion seem to be important routes of elimination for a number of the newer fluoroquinolones, such as grepafloxacin (GPFX), moxifloxacin (MFLX), and trovafloxacin (Emmerson and Jones, 2003). Urinary recovery of unchanged drug was high (>75% of dose) for levofloxacin (LVFX) and gatifloxacin, and was low (<20% of dose) for GPFX, MFLX, and trovafloxacin (Lubasch et al., 2000).
The fluoroquinolones have a carboxylic acid moiety at the C-3 position of the base molecule. Therefore, many fluoroquinolones are metabolized to their acyl glucuronides in humans. For example, it has been reported that the urinary excretion of acyl glucuronides accounted for 27 to 38% of the dose for sparfloxacin (Montay, 1996), 13.6% of the dose for MFLX (Stass and Kubitza, 1999), 12.8% of the dose for trovafloxacin (Dalvie et al., 1997), and 4.0% of the dose for GPFX (Akiyama et al., 1995). Glucuronidation of endogenous and xenobiotic substrates is catalyzed by UDP-glucuronosyltransferases (UGTs), which are mostly located in the endoplasmic reticulum of cells (Radominska-Pandya et al., 1999). Currently, 18 human UGT enzymes have been identified, and they have been classified into three subfamilies: UGT1A, 2A, and 2B (Mackenzie et al., 1997; Miners et al., 2004). Although the substrate specificities of UGTs are broad and overlapping, several classes of xenobiotic substrates are known to be glucuronidated by specific UGT enzymes (Tukey and Strassburg, 2000). Because little is known about the enzymatic basis of fluoroquinolone glucuronidation in humans, the goal of the present study was to determine which human UGT enzymes are responsible for the in vitro formation of the acyl glucuronides of LVFX, GPFX, and MFLX, and another new fluoroquinolone, sitafloxacin (STFX).

Chemical structures of fluoroquinolones.
Materials and Methods
Materials. LVFX hemihydrate, GPFX hydrochloride monohydrate, and STFX sesquihydrate were synthesized at Daiichi Pharmaceutical Co., Ltd. MFLX hydrochloride was purchased from LKT Laboratories (St. Paul, MN). Bilirubin and mefenamic acid were purchased from Sigma-Aldrich (St. Louis, MO). UDP-glucuronic acid, MgCl2, alamethicin, and all other reaction buffer components were purchased as a mix from BD Biosciences (San Jose, CA). Pooled human liver microsomes, individual donor human liver microsomes (HH13, HH18, HK23, HK25, HG32, HK37, HG42, HG43, HH47, HG56, HG64, HG74, HG93, and HG95), and recombinant human UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15, and 2B17 expressed in baculovirus-infected insect cells (Supersomes) were also purchased from BD Biosciences. All other chemicals and reagents were obtained from commercial suppliers and were of analytical grade.
Biosynthesis of LVFX, MFLX, GPFX, and STFX Acyl Glucuronides. A polyethylene cannula (PE10 connected to PE50; Becton, Dickinson and Company, Franklin Lakes, NJ) was surgically inserted into the common bile duct of male CD (SD) IGS rats (7 weeks of age, body weight 265–315 g; Charles River Japan Inc., Yokohama, Japan) that had been anesthetized with diethyl ether. After recovery from anesthesia, the bile duct-cannulated rats were given a single oral dose of LVFX, GPFX, MFLX, or STFX (100 mg/kg) suspended in a 0.5% w/v aqueous sodium carboxymethylcellulose solution (n = 3). Each rat was then placed into a separate Bollman cage, and bile samples were collected over 24 h in a container kept on ice. After the addition of 5 ml of 0.5 M acetate buffer (pH 5.0) to stabilize acyl glucuronides, the bile samples were stored at -30°C until use for purification of acyl glucuronides. All animals were allowed free access to pellet food and water, but they fasted overnight (ca. 16 h) before drug administration and until 4 h after administration. The study room was environmentally controlled for temperature (23°C ± 3°C), relative humidity (55% ± 15%), and light (a 12-h light/dark cycle). All animal studies conducted were approved by the institutional ethics committee prior to the study.
To purify the acyl glucuronides, bile samples were pooled by compound administered and centrifuged (1700g for 10 min at 4°C), and each supernatant was separately loaded onto a Mega Bond Elut C18 column (2 g/10 ml; Varian, Inc., Palo Alto, CA), which had been preconditioned with water and acetonitrile followed by 0.5 M acetate buffer (pH 5.0). The column was then washed with water (20 ml) and successively eluted with 20 ml of 10% and 20% v/v acetonitrile in 0.2% v/v aqueous acetic acid. Each eluate was evaporated to dryness under reduced pressure, and the residues were separately dissolved in 0.2% v/v aqueous acetic acid. Then, the preparative HPLC was used to purify the acyl glucuronide of each fluoroquinolone (see HPLC Analysis). The acyl glucuronide fraction was concentrated, desalted using a solid-phase extraction on a Mega Bond Elut C18 column (1 g/6 ml; Varian, Inc.), and lyophilized. 1H NMR and MS were used to determine the structures of purified acyl glucuronides (Fig. 1), and the results are as follows.
LVFX Glucuronide.1H NMR (D2O): δ 8.73 (1H, s, H-2), 7.32 (1H, d, J = 12.0 Hz, H-5), 5.68 (1H, d, J = 7.8 Hz, H-1′″), 4.43 (1H, d, J = 11.0 Hz, H-2′a), 4.32 (1H, d, J = 9.6 Hz, H-2′b), 3.91 (1H, d, J = 9.2 Hz, H-5′″), 3.62 to 3.53 (3H, m, H-2″′, H-3″′, and H-4″′), 3.51 to 3.44 (6H, m, H-2″, H-3″a, H-5″a and H-6″), 3.22 to 3.17 (2H, m, H-3″b and H-5″b), 2.85 (3H, s, 4″-methyl), 1.36 (3H, d, J = 6.9 Hz, 3′-methyl), MS [electrospray ionization (ESI)]: m/z 538 [M + H]+.
GPFX Glucuronide.1H NMR (D2O): δ 8.67 (1H, s, H-2), 7.30 (1H, d, J = 5.7 Hz, H-8), 5.66 (1H, d, J = 7.4 Hz, H-1″′), 3.88 (1H, d, J = 9.2 Hz, H-5″′), 3.64 (2H, d, J = 13.7 Hz, H-2″a and H-6″a), 3.58 to 3.40 (6H, m, H-1′, H-3″, H-5″a, H-2″′, H-3″′, and H-4″′), 3.30 to 3.21 (1H, m, H-5″b), 3.08 (1H, t, J = 12.6 Hz, H-6″b), 2.92 to 2.86 (1H, m, H-2″b), 2.49 (3H, s, 5-methyl), 1.27 (3H, t, J = 6.0 Hz, 3″-methyl), 1.20 (2H, d, J = 7.4 Hz, H-2′a and H-3′a), 0.97 to 0.95 (2H, m, H-2′b and H-3′b), MS (ESI): m/z 535 [M + H]+.
MFLX Glucuronide.1H NMR (D2O): δ 8.83 (1H, s, H-2), 7.53 (1H, d, J = 14.2 Hz, H-5), 5.69 (1H, d, J = 7.3 Hz, H-1″′), 4.04 to 3.99 (1H, m, H-1′), 3.98 (1H, d, J = 4.6 Hz, H-9″a), 3.90 (1H, d, J = 9.6 Hz, H-5″′), 3.82 (1H, t, J = 5.0 Hz, H-8″), 3.77 to 3.67 (2H, m, H-6″), 3.60 to 3.48 (4H, m, H-9″b, H-2″′, H-3″′, and H-4″′), 3.46 (3H, s, 8-methoxy), 3.29 (1H, d, J = 11.9 Hz, H-2″a), 2.96 (1H, t, J = 10.8 Hz, H-2″b), 2.71 (1H, broad s, H-3″), 1.81 to 1.71 (4H, m, H-4″ and H-5″), 1.12 to 0.78 (4H, m, H-2′ and H-3′), MS (ESI): m/z 578 [M + H]+.
STFX Glucuronide.1H NMR (D2O): δ: 8.83 (1H, d, J = 2.7 Hz, H-2), 7.69 (1H, d, J = 13.3 Hz, H-5), 5.70 (1H, d, J = 7.3 Hz, H-1″′), 4.90 (1H, ddd, J = 63.7, 9.2, 5.5 Hz, H-2′), 4.28 (1H, td, J = 7.4, 4.0 Hz, H-2″a), 4.15 to 4.12 (1H, m, H-1′), 4.04 (1H, d, J = 10.1 Hz, H-7″a), 3.91 (1H, d, J = 9.2 Hz, H-5″′), 3.60 (1H, d, J = 11.5 Hz, H-2″b), 3.60 to 3.50 (3H, m, H-2″′, H-3″′, and H-4″′), 3.41 (1H, d, J = 4.6 Hz, H-3″), 3.10 (1H, d, J = 9.6 Hz, H-7″b), 1.54 to 1.46 (1H, m, H-3′a), 1.21 (1H, d, J = 9.6 Hz, H-3′b), 0.93 to 0.71 (4H, m, H-5″ and H-6″), MS (ESI): m/z 586 [M + H]+.
In Vitro Glucuronidation Assay. Each reaction mixture (500 μl) contained 25 mM Tris-HCl buffer, pH 7.4, 5 mM MgCl2, 3 mM UDP-glucuronic acid, 25 μg/ml alamethicin, human liver microsomes or recombinant human UGT isoform (0.5 mg of protein/ml), and the substrate. The reaction was started by the addition of each substrate solution (10 μl in 2% v/v aqueous acetic acid), and each reaction mixture was incubated at 37°C for 60 min. The reactions were terminated by the addition of 1 ml of 2% v/v aqueous acetic acid in acetonitrile followed by cooling on ice. The internal standard (Table 1) was then added to each incubate, the samples were centrifuged (12,000g for 10 min at 4°C), and each supernatant was evaporated to dryness under nitrogen stream at room temperature. Each residue was then separately reconstituted with 200 μl of the initial mobile phase (Table 1) and filtered through an Ultrafree-MC PVDF membrane (0.45-μm mesh; Millipore Corporation, Billerica, MA). Finally, a 40-μl aliquot of each solution was loaded onto the HPLC apparatus.
HPLC conditions used to determine enzymatic fluoroquinolone glucuronidation
HPLC Analysis. A Hitachi D-7000 system consisting of two L-7000 pumps, an L-7200 autosampler, an L-7300 column oven, and an L-7480 fluorescence detector (Hitachi, Ibaraki, Japan) was used to conduct the HPLC analyses. To purify the acyl glucuronides from the bile extracts, separation was achieved with a Symmetry C18 column (5 μm, 7.6 × 100 mm; Waters, Milford, MA) at 40°C. Mobile phase A consisted of 0.2% aqueous acetic acid/acetonitrile (95:5 v/v) and mobile phase B consisted of 0.2% aqueous acetic acid/acetonitrile (50:50 v/v). At a constant flow rate of 3 ml/min, the LVFX glucuronide was eluted by the isocratic condition of 100% mobile phase A; the GPFX, MFLX, and STFX glucuronides were eluted by the isocratic condition of 70% mobile phase A. The UV detection set at 295 nm was monitored to collect the fractions that contain the acyl glucuronide. Each acyl glucuronide was further purified on the same column; an isocratic elution with a mobile phase consisting of 25 mM ammonium formate (pH adjusted to 3 with formic acid) and acetonitrile at a flow rate of 4 ml/min was used to achieve the separation. The LVFX glucuronide was eluted with 5% acetonitrile, the GPFX glucuronide was eluted with 10% acetonitrile, and the MFLX and STFX glucuronides were eluted with 12% acetonitrile.
To determine the concentration of glucuronide in the reaction mixture, quantitative separation was achieved using a Symmetry Shield RP18 column (3.5 μm, 4.6 × 150 mm; Waters) equipped with a Sentry guard column (Symmetry Shield RP18, 3.5 μm, 3.9 × 20 mm; Waters) at 40°C. The flow rate was 1 ml/min. Mobile phase A was 50 mM potassium phosphate buffer (pH adjusted to 2.0 with orthophosphoric acid) and mobile phase B was tetrahydrofuran. The separation and detection conditions are summarized in Table 1. The biosynthesized acyl glucuronides were dissolved in 2% v/v aqueous acetic acid and were used as the calibration standards. The calibration curves were generated using a weighed (1/concentration) least-squares linear regression of the peak area ratios (calibration standard/internal standard) plotted against the prepared concentrations.
NMR and Mass Spectrometry of Acyl Glucuronides.1H NMR spectra were recorded on a JNM-ECA500 (JEOL, Tokyo, Japan) operating at 500 MHz. Samples were dissolved in deuterium oxide containing 0.9% v/v acetic acid-d4 (Euriso-Top, Saint-Aubin, France). Chemical shifts were referenced internally to residual HDO in the solvent at δ 4.65 ppm. Mass spectrometry was performed using an ion-trap LCQ spectrometer (Thermo Electron Corporation, Waltham, MA). ESI was operated in positive mode. The capillary temperature was set at 200°C, and the ion spray voltage was set at 4.5 kV. Each sample (ca. 2 μg/ml), dissolved in 0.2% v/v aqueous acetic acid/acetonitrile (50:50 v/v), was introduced into the mass spectrometer at a constant flow rate of 8 μl/min.
Determination of Kinetic Constants. Enzyme kinetics of the glucuronidation by pooled human liver microsomes and the recombinant human UGT1A1 and 1A9 enzymes was investigated at various fluoroquinolone concentrations (LVFX, 50–4000 μM; GPFX and STFX, 10–2500 μM; MFLX, 10–2000 μM). The apparent Michaelis constant (Km) and maximum velocity (Vmax) were determined by nonlinear regression analysis using GraFit 5.0 software (Erithacus Software, Horley, Surrey, UK), assuming Michaelis-Menten kinetics over the range of substrate concentration tested. The formation rates of acyl glucuronide (v) for each substrate concentration [S] were fit to the following equation: v = Vmax × [S]/(Km + [S]).
Activity Screen with Recombinant Human UGT Enzymes. LVFX, GPFX, MFLX, and STFX were separately incubated with 12 commercially available recombinant human UGT-expressing microsomes. The membrane preparation from insect cells infected with wild-type baculovirus (BD Biosciences) was used as a negative control. Two substrate concentrations (100 μM and 2 mM) were used in this study; the higher concentration was selected by approximating the Km values for human liver microsomes, and the lower concentration was approximately 10-fold higher than the clinically relevant plasma concentrations of STFX (O'Grady et al., 2001), because the concentrations of 14C-STFX-related radioactivity in rat liver were 8- to 13-fold of those in the serum (Tachibana et al., 2004).
Correlation Analysis. The glucuronidation activities of LVFX, GPFX, MFLX, and STFX at the concentration of 100 μM were measured in a bank of human liver microsomes from 14 individual donors and then compared with UGT-selective marker activities. Pearson's product-moment correlation coefficient (r) was used to assess the relationship between glucuronidation activities for each fluoroquinolone and UGT1A1-selective β-estradiol 3-glucuronidation activity (Senafi et al., 1994), UGT1A4-selective trifluoperazine glucuronidation activity (Green and Tephly, 1995), and UGT1A9-selective propofol glucuronidation activity (Burchell et al., 1995). The data on individual UGT enzyme activity in each sample were provided by the manufacturer. The computer program JMP 5.1 (SAS Institute, Cary, NC) was used to conduct the correlation analysis, and a P < 0.05 for the correlation coefficient (r) was considered statistically significant.
Chemical Inhibition Study. The glucuronidation activities of LVFX, GPFX, MFLX, and STFX at the concentration of 100 μM were measured in pooled human liver microsomes in the presence of known UGT inhibitors. The chemical inhibitors include bilirubin for UGT1A1 (Williams et al., 2002) and mefenamic acid for UGT1A9 (McGurk et al., 1996; Wynalda et al., 2003). Both inhibitors were dissolved in dimethyl sulfoxide and were added to the reaction mixtures at the final concentrations of 1 to 500 μM. Control incubations containing all components of the reaction mixtures, including 1% v/v dimethyl sulfoxide, but not the inhibitors, were performed in parallel. The glucuronidation activities were calculated as a percentage of control activity, and the IC50 values were estimated by nonlinear regression analysis using GraFit 5.0 software with the following equation (Houston et al., 2003): % of control = [Range/(1 + [I]/IC50)s] + Background, where [I] is the inhibitor concentration, Range is the fitted uninhibited value minus the Background, and s is a slope factor.

Representative chromatograms of reaction mixtures after incubation of LVFX (A), GPFX (B), MFLX (C), and STFX (D) with human liver microsomes in the presence of UDP-glucuronic acid. The concentration of each substrate was 100 μM. Separation and detection conditions were described under Materials and Methods and Table 1. IS is the internal standard.
Results
Glucuronidation of Fluoroquinolones by Pooled Human Liver Microsomes. Representative chromatograms of reaction mixtures containing a 100 μM concentration of each substrate are shown in Fig. 2. Because GPFX is a racemic mixture of R(+)- and S(-)-isomers, the reaction yields two glucuronide stereoisomers (Fig. 2B). As shown in Fig. 3, the formation rate of each fluoroquinolone glucuronide is concentration-dependent. Nonlinear regression of the data yielded the apparent Km values, ranging from 1.9 mM to 10.0 mM (Table 2). It should be noted that the Km values that exceed the highest concentration of substrate are provided as rough estimates. The apparent Vmax/Km values are highest for MFLX (0.12 μl/min/mg protein), moderate for STFX (0.048 μl/min/mg protein) and GPFX (0.022 μl/min/mg protein), and lowest for LVFX (0.0028 μl/min/mg protein).
Apparent kinetic parameters for the glucuronidation of fluoroquinolones by human liver microsomes For Km and Vmax values, glucuronidation activities from triplicate incubations were averaged and nonlinear regression was used to fit the data to the Michaelis-Menten equation. The parameter estimated and the S.E. value generated by the GraFit 5.0 software are shown.
Correlation Studies with Human Liver Microsomes from Individual Donors. As shown in Fig. 4, high and statistically significant correlations occurred between UGT1A1-selective β-estradiol 3-glucuronidation activity and LVFX (r = 0.823, P = 0.0003), MFLX (r = 0.706, P = 0.005), and STFX (r = 0.756, P = 0.002) glucuronidation activities. In contrast, GPFX glucuronidation activity correlated highly with UGT1A9-selective propofol glucuronidation activity (r = 0.787, P = 0.0008). Relatively weak, but statistically significant correlations were observed between STFX and LVFX glucuronidation and propofol glucuronidation; a weak correlation was also observed between GPFX glucuronidation and β-estradiol 3-glucuronidation (Table 3). It is important to note that, in a bank of human liver microsomes from 14 individual donors, significant correlation was found between β-estradiol 3-glucuronidation and propofol glucuronidation (r = 0.621, P = 0.0178). Therefore, a weak background correlation would be observed because of coregression. There was no correlation between fluoroquinolone glucuronidation and trifluoperazine glucuronidation, which is a marker of UGT1A4 activity.
Correlation coefficients between UGT-selective catalytic activities and fluoroquinolone glucuronidation activities by human liver microsomes from 14 individual donors
Glucuronidation of Fluoroquinolones by Recombinant Human UGTs. At both low (100 μM) and high (2 mM) substrate concentrations, the glucuronidation of LVFX, GPFX, MFLX, and STFX was catalyzed by UGT1A1, 1A3, 1A7, and 1A9 enzymes (Fig. 5). Other UGT1A enzymes (1A4, 1A5, 1A6, 1A8, and 1A10) and 2B enzymes (2B4, 2B7, 2B15, and 2B17; data not shown) did not catalyze the glucuronidation of the fluoroquinolones examined as part of this study.
Enzyme Kinetics of Fluoroquinolone Glucuronidation by the Recombinant Human UGT1A1 and 1A9 Enzymes. The formation rate of each fluoroquinolone glucuronide by the recombinant human UGT1A1 and 1A9 was investigated further. All reactions followed typical Michaelis-Menten kinetics (data not shown). As shown in Table 4, UGT1A1 and 1A9 showed high Km values (>1 mM) for all reactions. The results showed that UGT1A1 most efficiently glucuronidates MFLX, whereas UGT1A9 most efficiently glucuronidates GPFX.
Apparent kinetic parameters for the glucuronidation of fluoroquinolones by recombinant UGT1A1 and UGT1A9 enzymes For Km and Vmax values, glucuronidation activities from triplicate incubations were averaged and nonlinear regression was used to fit the data to the Michaelis-Menten equation. The parameter estimated and the S.E. value generated by the GraFit 5.0 software are shown.
Chemical Inhibition Study. The inhibition of the glucuronidation activities of LVFX, GPFX, MFLX, and STFX was attempted using UGT isoform-selective chemical inhibitors, bilirubin (UGT1A1) and mefenamic acid (UGT1A9), in human liver microsomes. As shown in Fig. 6, bilirubin exhibited a potent inhibition of the glucuronidation activities of MFLX and STFX. The IC50 values were 4.9 μM for MFLX and 4.7 μM for STFX. The glucuronidation of LVFX was also inhibited by bilirubin (IC50 = 3.1 μM), but approximately 40% of the activity remained uninhibited. The inhibitory effect of bilirubin on the glucuronidation of GPFX was relatively weak, although approximately 45% of the activity was inhibited. In contrast, mefenamic acid showed a potent inhibition of the glucuronidation activity of GPFX with an IC50 of 4.9 μM; however, the inhibitory effects of mefenamic acid on the glucuronidation of other fluoroquinolones were relatively weak (IC50 > 50 μM).

Enzyme kinetics of LVFX (A), GPFX (B), MFLX (C), and STFX (D) glucuronidation by human liver microsomes. Each value represents the mean ± S.E. of triplicate incubations. The lines are computer-fitted curves resulting from nonlinear regression analysis. Insets are Eadie-Hofstee plots.
Discussion
Fluoroquinolone antibiotics have been widely used to treat urinary and respiratory tract infections for more than 40 years. Although many fluoroquinolones, such as sparfloxacin (Montay, 1996) and MFLX (Stass and Kubitza, 1999), are metabolized to their acyl glucuronides, there has been no study that tried to characterize the enzyme kinetic properties of this metabolic reaction. Hence, in the present study, biosynthesised acyl glucuronides were purified from rat bile, and were used to develop HPLC analytical methods for the determination of glucuronides produced by in vitro microsomal reactions. The formation of LVFX, GPFX, MFLX, and STFX acyl glucuronides was then investigated using human liver microsomes and recombinant human UGT enzymes expressed by baculovirus-infected insect cells.
For the pooled human liver microsomes, the estimated intrinsic clearance values (measured as Vmax/Km) suggest that MFLX is the best substrate for human UGTs, followed by STFX and GPFX. The results also indicate that LVFX undergoes only limited glucuronidation in human liver microsomes. In clinical studies conducted in healthy male volunteers, the urinary excretion of the acyl glucuronide accounted for 13.6% of the dose for MFLX (Stass and Kubitza, 1999) and 4.0% of the dose for GPFX (Akiyama et al., 1995). In contrast, the LVFX glucuronide has not been identified in humans, because LVFX is mainly excreted as the unchanged form in the urine (Fish and Chow, 1997). The rank order of the Vmax/Km values (MFLX > GPFX >> LVFX) obtained from our studies is in good agreement with these clinical metabolism data.
Although the substrate specificities of UGTs are broad and overlapping, several classes of xenobiotic substrates are known to be glucuronidated by specific UGT enzymes. For example, bilirubin glucuronidation is selectively catalyzed by UGT1A1 (Bosma et al., 1994), propofol glucuronidation appears to be catalyzed by UGT1A9 (Burchell et al., 1995), and morphine 6-glucuronidation is selectively catalyzed by UGT2B7 (Coffman et al., 1997; Stone et al., 2003). In vitro activity screens with 12 different UGT enzymes revealed that UGT1A1, 1A3, 1A7, and 1A9 are capable of glucuronidating LVFX, GPFX, MFLX, and STFX (Fig. 4). Since UGT1A7 is not expressed in human liver (Strassburg et al., 1997), it is thought that UGT1A7 does not contribute to the hepatic glucuronidation of these fluoroquinolones. In addition, with the exception of LVFX, the glucuronidation activities by UGT1A3 are relatively low. Taken together with results from Northern blot analysis of human liver tissue samples that indicate that the expression of UGT1A3 was 20-fold less than that of UGT1A1 (Owens and Ritter, 1995; Mojarrabi et al., 1996), these results suggest that UGT1A3 has minor significance in the glucuronidation of fluoroquinolones in human liver microsomes.

Correlation plots of UGT isoform-selective catalytic activities and fluoroquinolone glucuronidation activities in human liver microsomes from 14 individual donors. (A) β-Estradiol 3-glucuronidation versus LVFX glucuronidation, (B) β-estradiol 3-glucuronidation versus MFLX glucuronidation, (C) β-estradiol 3-glucuronidation versus STFX glucuronidation, (D) propofol glucuronidation versus GPFX glucuronidation.
Interestingly, the results of activity screens and enzyme kinetics analysis with recombinant UGT enzymes show that UGT1A1 is the most efficient isoform for the glucuronidation of MFLX, but UGT1A9 is the most efficient isoform for the glucuronidation of GPFX (Fig. 5; Table 4). In addition, the results of correlation analysis using a bank of human liver microsomes (n = 14) show that the glucuronidation activities of LVFX, MFLX, and STFX correlate highly with UGT1A1-selective β-estradiol 3-glucuronidation activity, but the glucuronidation activity of GPFX correlates highly with UGT1A9-selective propofol glucuronidation activity (Table 3). Moreover, the UGT1A1-selective inhibitor bilirubin exhibited a potent inhibition of the glucuronidation of MFLX and STFX in human liver microsomes (Fig. 6). In contrast, the proposed UGT1A9 inhibitor, mefenamic acid, exhibited a greater inhibition of the glucuronidation of GPFX compared with the other fluoroquinolones. These results suggest that 1) UGT1A1 predominantly catalyzes the glucuronidation of MFLX and STFX, 2) UGT1A9 mainly catalyzes the glucuronidation of GPFX, and 3) both UGT1A1 and 1A9 are involved in the glucuronidation of LVFX in human liver microsomes.
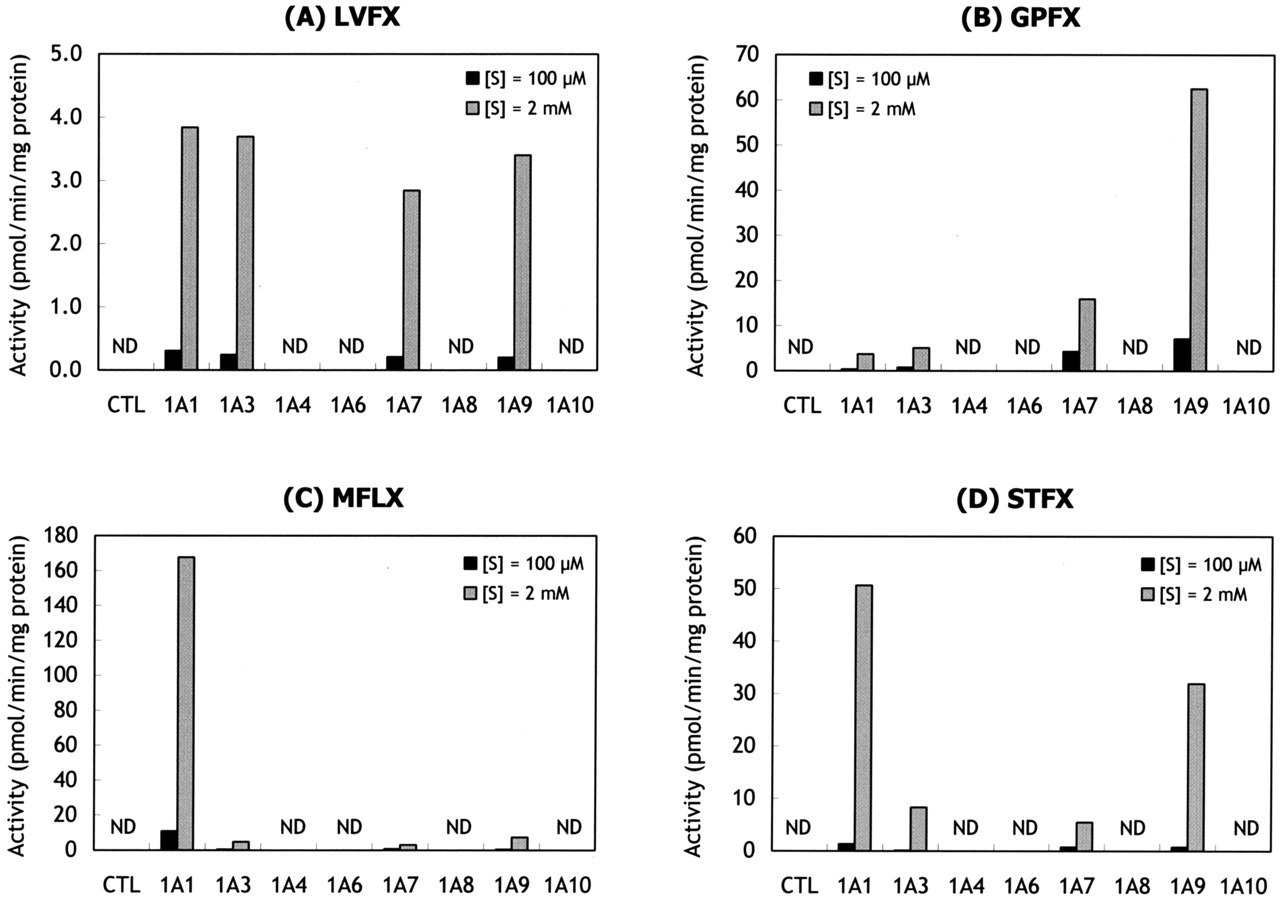
Glucuronidation of LVFX (A), GPFX (B), MFLX (C), and STFX (D) by recombinant human UGT enzymes. Each substrate (100 μM or 2 mM) was incubated with human UGT control microsomes (CTL) or recombinant UGT enzymes at 37°C for 60 min. Each value represents the mean of duplicate incubations. ND, not detected.
UGT1A1 is a predominant isoform for the acyl glucuronidation of bilirubin (Bosma et al., 1994). However, there are few examples of the formation of acyl glucuronides by UGT1A1 because other carboxylic acids, including nonsteroidal anti-inflammatory drugs, are not glucuronidated by this isoform (Tukey and Strassburg, 2000). It has been reported that acyl glucuronidation of the endothelin ETA receptor antagonist compound A was catalyzed by several UGT enzymes including UGT1A1 (Tang et al., 2003), but the contribution of this enzyme was relatively minor. The results of the present study clearly show that MFLX glucuronidation is mainly catalyzed by UGT1A1. This is the first report that describes a major role of UGT1A1 for acyl glucuronidation of carboxylic acid-containing drugs in human liver microsomes.
Many carboxylic acid-containing drugs, such as nonsteroidal anti-inflammatory drugs, clofibric acid, and valproic acid, have been reported to be glucuronidated by UGT2B7, which is a major isoform in both liver and gastrointestinal tissue (Jin et al., 1993; King et al., 2001). However, the results of the present study show that all the UGT2B enzymes, including UGT2B7, do not catalyze the glucuronidation of fluoroquinolones. Recently, Sakaguchi et al. (2004) described that phenyl or biphenyl compounds bearing carboxylic acid groups with additional functional groups such as fluorines or chlorines decrease the rate of glucuronidation by UGT2B7. In addition, the glucuronidation of coumarin 3-carboxylic acid has not been detected by UGT2B7 (Sakaguchi et al., 2004). Thus, the fluoroquinolones are unlikely to be good substrates for UGT2B7 because they possess a fluorine atom at the C-6 position of the 3-carboxylic acid quinolone molecule.

Effects of chemical inhibitors on the glucuronidation of LVFX, GPFX, MFLX, and STFX in human liver microsomes. Each substrate (100 μM) was incubated with human liver microsomes in the presence of bilirubin (A) or mefenamic acid (B) at 37°C for 60 min. The final concentrations of each inhibitor in the reaction mixtures ranged from 1 to 500 μM. Each value represents the mean of duplicate incubations.
Acyl glucuronides are the major metabolites of many carboxylic acid-containing drugs and have been identified as reactive electrophilic metabolites (Spahn-Langguth and Benet, 1992). The chemical reactivity of acyl glucuronides has been well established; results of other studies show that they can undergo hydrolysis or intramolecular acyl migration, and may react with proteins leading to covalent drug-protein adducts (Bailey and Dickinson, 2003). Some of these drugs, including benoxaprofen, tolmetin, zomepirac, and diflunisal, cause toxic idiosyncratic reactions probably related to the formation of covalent adducts between the reactive acyl glucuronide and a functional protein (Shipkova et al., 2003). Among the fluoroquinolone-related compounds, the specific idiosyncratic adverse reactions have been reported with the 1-(2,4)-difluorophenyl quinolones, such as temafloxacin-associated hemolytic uracemic-like syndrome and trovafloxacin-associated liver failure (Ball, 2003). Andersson and MacGowan (2003) hypothesized that metabolites of these agents, which share common structures, may be responsible for some of the tissue-specific adverse reactions. Additional studies are needed to further elucidate the relationship between the chemical reactivity of acyl glucuronides of fluoroquinolones and the immunologically mediated adverse reactions.
In conclusion, the present study identified that UGT1A1, 1A3, and 1A9 enzymes are involved in the acyl glucuronidation of LVFX, GPFX, MFLX, and STFX in human liver microsomes. Furthermore, the results of correlation analysis and the UGT-selective inhibition study suggested that MFLX and STFX are mainly glucuronidated by UGT1A1, and that GPFX is mainly glucuronidated by UGT1A9. Since newer fluoroquinolones seem to be eliminated by metabolism (Andersson and MacGowan, 2003), information on which UGT enzymes are primarily responsible for glucuronidation is necessary to understand potential pharmacokinetic variability and to predict potential drug interactions.
Acknowledgments
We thank Steven E. Johnson for editing the manuscript.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.003178.
-
ABBREVIATIONS: GPFX, grepafloxacin; LVFX, levofloxacin; MFLX, moxifloxacin; STFX, sitafloxacin; UGT, UDP-glucuronosyltransferase; MS, mass spectrometry; ESI, electrospray ionization; HPLC, high-performance liquid chromatography; compound A, (+)-(5S,6R,7R)-2-isopropylamino-7-[4-methoxy-2-((2R)-3-methoxy-2-methylpropyl)]-5-(3,4-methylenedioxyphenyl)cyclopenteno[1,2-b]pyridine 6-carboxylic acid.
- Received November 29, 2004.
- Accepted March 14, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}