


Abstract
Knowledge of drug absorption, distribution, metabolism, and excretion (ADME) or pharmacokinetics properties is essential for drug development and safe use of medicine. Varied or altered ADME may lead to a loss of efficacy or adverse drug effects. Understanding the causes of variations in drug disposition and response has proven critical for the practice of personalized or precision medicine. The rise of noncoding microRNA (miRNA) pharmacoepigenetics and pharmacoepigenomics has come with accumulating evidence supporting the role of miRNAs in the modulation of ADME gene expression and then drug disposition and response. In this article, we review the advances in miRNA pharmacoepigenetics including the mechanistic actions of miRNAs in the modulation of Phase I and II drug-metabolizing enzymes, efflux and uptake transporters, and xenobiotic receptors or transcription factors after briefly introducing the characteristics of miRNA-mediated posttranscriptional gene regulation. Consequently, miRNAs may have significant influence on drug disposition and response. Therefore, research on miRNA pharmacoepigenetics shall not only improve mechanistic understanding of variations in pharmacotherapy but also provide novel insights into developing more effective therapeutic strategies.
Introduction
The utility of a drug depends on its efficacy and safety profiles. Upon entering the body, the drug is subjected to absorption, distribution, metabolism, and excretion (ADME) processes before acting on its molecular target to exert pharmacological or toxicological effects. Change in ADME may lead to variable levels of drug for target binding and consequently have significant impact on drug efficacy and safety profiles, which could cause a reduction/loss of pharmacological effects or adverse events (Lu, 1998; Haga et al., 2006; Giacomini et al., 2010; Yu and Pan, 2012). Therefore, research on ADME processes and causes of variation is essential for developing better drugs and ensuring the safe use of approved medications.
ADME processes are mechanistically controlled by drug-metabolizing enzymes and transporters expressed in various tissues including small intestine, liver, and kidney. Drug-metabolizing enzymes consist of Phase I [e.g., cytochrome P450 (CYP or P450)] and Phase II [e.g., uridine 5′-diphospho-glucuronosyltransferase (UGT)] enzymes that are able to convert the drug to a more hydrophilic and polar metabolite and determine hepatic drug clearance. Transporters including ATP binding cassette (ABC) and solute carrier (SLC) proteins mediate the transport (e.g., efflux or uptake) of many drugs and may have significant effects on drug absorption, distribution, and excretion processes. Therefore, change in drug-metabolizing enzyme and transporter gene expression or protein activity would ultimately alter ADME or pharmacokinetics properties and subsequently affect therapeutic outcomes.
Many mechanisms behind variable ADME have been discovered, which may help to develop more rational and improved therapeutics. For instance, genetic variations can have significant impact on the expression or function of drug-metabolizing enzymes (e.g., CYP2D6 and UGT1A1) and transporters (e.g., ABCB1) and consequently alter drug disposition and response. Therefore, doses may be adjusted or an alternative drug may be prescribed for patients with particular high-risk genotypes or phenotypes, namely personalized or precision medicine, to achieve the desired efficacy and prevent adverse effects. Furthermore, drug-metabolizing enzyme and transporter gene expression is regulated by nuclear receptors [NRs; e.g., pregnane X receptor (PXR or NR1I2)) and transcription factors, and modulated through signal transduction, posttranslational modification, membrane trafficking and subcellular organization pathways (for reviews, see Correia and Liao, 2007; Morgan, 2009; Gu and Manautou, 2010; Klaassen and Aleksunes, 2010; Tolson and Wang, 2010)]. Activation or suppression of such regulatory factors or pathways would cause significant change in enzyme/transporter levels and activities and lead to multidrug resistance (MDR), loss of efficacy, or adverse drug effects. Knowledge of these mechanisms has proven helpful for the prediction and prevention of possible toxicity risks and the development of more effective and safer treatments.
Increased research on pharmacoepigenetics and pharmacoepigenomics has demonstrated the role of epigenetic factors in controlling ADME gene expression, in particular, by noncoding microRNAs (miRNAs or miR), DNA methylation proteins, and histone modification proteins (see recent reviews, Ivanov et al., 2012; Ingelman-Sundberg et al., 2013; Zhong and Leeder, 2013). A number of studies have shown that methylation of cytosine-phosphate-guanine sites located in the promoter regions of ADME genes or acetylation of histones may alter ADME gene expression in cells. In addition, there is growing evidence that miRNAs may modulate cellular ADME processes through posttranscriptional regulation of ADME gene expression (Fig. 1). In this review, we briefly introduce the general characteristics of noncoding miRNAs and focus on the discussion of miRNA-controlled epigenetic regulation of drug metabolism and disposition.

Role of miRNAs in the regulation of drug metabolism and transport. MicroRNAs may modulate cellular drug metabolism and transport capacity through targeting of drug-metabolizing enzymes, transporters, xenobiotic receptors, and/or other regulatory factors whereas the biogenesis of miRNAs may be regulated by xenobiotic receptors or other regulatory factors.
MicroRNAs in Posttranscriptional Gene Regulation
MicroRNAs are short, genome-derived, noncoding RNAs (ncRNAs) that exhibit cell specific expression profiles during development and in response to xenobiotics or other environmental factors. Since the discovery of the first miRNA lin-4 in Caenorhabditis elegans in 1993, more than 1800 miRNA sequences have been identified in humans and predicted to regulate thousands of protein-coding genes essentially modulating all cellular processes.
Biogenesis of miRNAs.
Mature miRNAs are produced within cells via canonical or alternative biogenesis pathways. The canonical pathway is dependent upon both DiGeorge syndrome critical region 8 (Drosha-DGCR8) and Dicer-TRBP (transactivation-response RNA-binding protein) complexes (Fig. 2), whereas the noncanonical pathway is not (Yang and Lai, 2011). Canonical biogenesis starts from the transcription of miRNA-coding sequences by RNA polymerase II to long primary miRNA (pri-miRNA) transcripts. Drosha-DGCR8 then cleaves pri-miRNAs to shorter hairpin precursor miRNAs (pre-miRNAs) within the nucleus. After being transported to the cytoplasm by Exportin-5, pre-miRNAs are cleaved by the Dicer-TRBP complex into double-stranded miRNA duplexes, which upon unwinding produce single-stranded mature miRNAs (Fig. 2). By contrast, noncanonical miRNAs are generated from short hairpin introns (mirtrons) by splicing machinery and lariat-debranching enzyme (Curtis et al., 2012) or derived from particular hairpin ncRNA species (e.g., small nucleolar RNAs or transfer RNAs) by other RNases (Cole et al., 2009; Babiarz et al., 2011), rather than Drosha-DGCR8 and Dicer-TRBP.

The canonical pathway of miRNA biogenesis and the mechanisms of miRNA controlled posttranscriptional gene regulation. The miRNA coding sequences are transcribed by RNA polymerase II to pri-miRNAs in the nucleus, which are then processed by the Drosha-DGCR8 complex to pre-miRNAs. Exportin 5 transports pre-miRNAs into cytoplasm, where pre-miRNAs are further processed by the Dicer-TRBP complex to double-stranded miRNAs. The miRNA duplexes are subsequently unwound, and the single-stranded mature miRNAs are loaded into argonaute 2-containing miRISCs. As discussed in the main text, some miRNAs may be generated through a number of different noncanonical pathways. By pairing to the MREs that usually occur within the 3′UTRs of target mRNAs, miRNAs mediate translational repression or mRNA degradation, which both may lead to lower levels of protein expression.
Mechanisms of miRNA-Controlled Posttranscriptional Gene Expression.
Once loaded into the argonaute 2-containing miRNA-induced silencing complexes (miRISCs) in the cytoplasm, miRNAs act on target transcripts via complementary Watson-Crick base pairing to corresponding miRNA response elements (MREs; Fig. 2), which are usually present within the 3′-untranslated regions (3′UTRs) of target genes. Upon binding to MRE sites, miRNAs reduce protein outcome from target transcripts, which may involve translational repression and/or mRNA deadenylation and decay mechanisms (Fig. 2). Although there had been some confusion about the contribution and timing of miRNA-induced translational inhibition and mRNA degradation effects, two recent in vivo studies clearly demonstrated that miRNAs largely inhibit translation (e.g., translation initiation) before causing mRNA decay (Bazzini et al., 2012; Djuranovic et al., 2012), emphasizing the importance of evaluating protein abundance rather than only examining mRNA levels for understanding miRNA-controlled posttranscriptional gene regulation.
Identification and Evaluation of microRNA Target Genes.
Based on the general principles of miRNA-target interactions, such as the strength of Watson-Crick complementarity, accessibility of MRE sites, and evolutionary conservation, a number of computational algorithms have been established for in silico prediction of putative MRE sites and targeted genes (for reviews, see Yu, 2007, 2009). Although computational prediction is helpful for the identification of possible miRNA targets for further investigation, there are unsurprisingly high levels of false positives, whereas other true targets are overlooked. In any case, biologic experiments are essential for the identification and verification of miRNA targets as well as delineation of the significance of miRNA-controlled gene regulation.
One experimental approach to screen miRNA targets is to examine the transcriptome of cells with lost or gained miRNA expression/function. Given the translational repression mechanism of miRNA-controlled gene regulation, many true miRNA targets may likely be overlooked by this method. Furthermore, other transcripts might be presented as false-positive targets that were actually altered as a result of transcriptional regulation or signal transduction caused by true miRNA targets. Another biologic approach is to directly pull down transcripts using the miRNA of interest or isolate argonaute 2 or miRISC complex-associated transcripts through immunoprecipitation and thus analyze those isolated mRNAs (and miRNAs) using various techniques including deep sequencing. This “gold standard” method relies largely on the stringency of experimental conditions, specificity of antibody for immunoprecipitation, and selectivity of other reagents. Given the fact that gene expression within live cells is very dynamic, experimental results may differ with experimental timing. Indeed this method presents many possible “targets”; however, this cannot be confirmed by other assays and the actual effects on protein outcome remain unknown. For this reason, proteomic approaches have become increasingly popular for the identification of possible miRNA targets in cells after the perturbation of miRNA expression or function (Vinther et al., 2006; Baek et al., 2008; Selbach et al., 2008; Leivonen et al., 2009a; Yang et al., 2009). Although proteomic approaches are able to directly define the effects of miRNAs on protein outcome and provide insight into miRNA targets, it is still unknown whether those altered proteins are direct or downstream effects of miRNA-controlled gene regulation.
Many methodologies have been developed for the validation of computationally predicted or experimentally identified miRNA target genes to delineate the significance of specific miRNAs in the control of cellular processes. Approaches include the evaluation of miRNA-MRE interactions (e.g., luciferase reporter assays with wild-type and mutant MRE sites, RNA electrophoresis mobility shift assay, etc.), assessment of relationship between miRNA and mRNA target expression (protein and/or mRNA levels), and examination of the impact of miRNA on target gene mRNA and protein expression as well as biologic functions. While one study (e.g., reporter assay) may offer some helpful information, the use of a combination of approaches is necessary to provide a more comprehensive understanding of miRNA-governed regulation and biologic significance. Furthermore, these studies often involve the use of both loss- and gain-of-miRNA-function strategies as well as in vitro and in vivo model systems (Yu, 2009; Yu and Pan, 2012). Gain-of-function approaches are commonly used in a system with minimal or no expression of miRNA, whereas loss-of-function approaches may be used in a model with excessive or regular levels of miRNA expression. Specifically, miRNA expression/function may be perturbed in cells or animals with miRNA mimics, precursors, antagomirs/inhibitors, sponges, and siRNAs as well as corresponding expression plasmids/viral vectors. In addition, some genetically modified animal models (Lu et al., 2007; Thai et al., 2007; Xiao et al., 2007; Han et al., 2015) have been developed and used for the examination of specific miRNA targets and functions at the whole organism level. Through the actions of target genes, miRNAs are thus able to control many cellular processes, including ADME, which may subsequently alter drug efficacy and safety profiles (Yu, 2009; Ivanov et al., 2012; Yu and Pan, 2012; Ingelman-Sundberg et al., 2013; Yokoi and Nakajima, 2013; Zhong and Leeder, 2013).
MicroRNAs Modulate Cellular Drug Metabolism Capacity through the Regulation of Phase I and II Enzyme Expression.
In an effort to address the gap in understanding posttranscriptional gene regulation mechanisms underlying variable drug metabolism, many studies have demonstrated that some CYP genes may be regulated by miRNAs in human cell line model systems (Table 1). Among them, a well-conserved miRNA miR-27b was shown to directly act on multiple CYPs including CYP1B1 and CYP3A4 (Tsuchiya et al., 2006; Pan et al., 2009a; Li et al., 2014b). Luciferase reporter assays were first employed to define the interactions of miR-27b with corresponding MRE sites within the 3′UTRs of CYP1B1 in MCF-7 and CYP3A4 in HEK-293 cells, which were predicted by bioinformatic algorithms. Other biologic experiments were conducted to elucidate the effects of miR-27b on CYP1B1 and CYP3A4 mRNA and protein expression as well as drug-metabolizing capacity (e.g., midazolam 1′-hydroxylation by endogenous CYP3A4) in various cell lines. The inverse relationship between miR-27b and CYP1B1 protein expression levels in human breast cancerous tissues (Tsuchiya et al., 2006) further demonstrated the significance of miR-27b-controlled regulation underlying variable CYP1B1 expression in cancerous tissues. Furthermore, the existence of miRNA-controlled posttranscriptional regulation mechanisms may help to understand developmental and sexual differences in CYP gene expression in humans and animals (for review, see Zhong and Leeder, 2013). Indeed the increase of hepatic miR-27b and murine mmu-miR-298 levels in mice after puberty (Pan et al., 2009a) may provide insight into the decreased or lost expression of CYP3A4 found in CYP3A4-transgenic mice (Granvil et al., 2003; Yu et al., 2005; Felmlee et al., 2008). It is also noteworthy that miR-27b/a was also shown to act on a number of ABC transporters (e.g., ABCB1; Table 2), xenobiotic receptors and transcription factors, e.g., vitamin D receptor (/NR1I1); Table 3 (see the following sections), suggesting that miR-27 may be an important miRNA related to cellular defense against xenobiotics.
MicroRNAs are shown to modulate the expression of Phase I and II drug-metabolizing enzymes
MicroRNAs are revealed to control the expression of efflux and uptake drug transporters
MicroRNAs are shown to regulate the expression of xenobiotic receptors or transcription factors
There are also many other experimentally evaluated CYP gene regulation by miRNAs (Table 1), including CYP1A1 by miR-892a (Choi et al., 2012), CYP2C8 by miR-103 and miR-107 (Zhang et al., 2012), CYP2C9 by miR-128-3p and miR-130b (Rieger et al., 2015; Yu et al., 2015a), CYP2E1 by miR-378 and miR-570 (Mohri et al., 2010; Nakano et al., 2015), CYP2J2 by let-7b (Chen et al., 2012a), and CYP24A1 by miR-125b (Komagata et al., 2009). The effects of miR-130b on endogenous CYP2C9 enzymatic activity (e.g., tolbutamide hydroxylation) in HepaRG cells were demonstrated by selective liquid chromatography-coupled tandem mass spectrometry assay (Rieger et al., 2015). Reporter assays further confirmed the direct action of miR-130b on CYP2C9 3′UTR. Interestingly, miR-130b also reduced the activities of all other measured CYPs by more than 30%, which was associated with significantly lower mRNA levels of constitutive androstane receptor (CAR/NR1I3), farnesoid X receptor, CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, and glutathione S-transferase alpha 2 (GSTA2). Because miR-130b levels are highly elevated in cholestatic liver and during inflammation (Rieger et al., 2013), these findings should advance molecular understanding of altered drug metabolism under pathophysiologic conditions.
Some Phase II drug-metabolizing enzymes, e.g., UGT1A1 and sulfotransferase 1A1 (SULT1A1), are posttranscriptionally regulated by specific miRNAs (Table 1). Computational analysis identified a putative miR-491-3p MRE site within the shared 3′UTR common to all UGT1A genes, which encode UGT1A enzymes responsible for the addition of glycuronosyl groups from uridine diphophosphate glucuronic acids onto drugs to form glucuronide metabolites. This miR-491-3p MRE was first verified using a luciferase reporter assay and mutagenesis studies (Dluzen et al., 2014). Furthermore, gain and loss of miR-491-3p function in Huh-7 cells led to a significant suppression and elevation of UGT1A1 mRNA levels and raloxifene glucuronidation activities, respectively. In addition, there was an inverse correlation between miR-491-3p and UGT1A3 as well as UGT1A6 mRNA levels among a set of normal human liver specimens, indicating that miR-491-3p may be an important epigenetic regulator in the control of hepatic UGT1A expression. In another study, SULT1A1, the most abundant hepatic SULT involved in the sulfation of various drugs, was revealed to be targeted by miR-631 in an allele specific manner (Yu et al., 2010), suggesting the importance of miR-631 pharmacoepigenetics among populations with particular SULT1A1 genotypes.
MicroRNAs Control Cellular Drug Transport Capacity via the Regulation of Transporter Expression.
Transporters mediate the translocation of many drugs (and endogenous compounds) across the cell membrane and consequently influence ADME processes as well as pharmacokinetic and dynamic properties. Some ABC transporters [e.g., P-glycoprotein (P-gp, MDR1 or ABCB1), breast cancer resistance protein (BCRP or ABCG2), and multidrug resistance-associated proteins (MRPs or ABCCs)] mainly function as efflux transporters and overexpression of such efflux transporter in MDR or diseased cells may confer MDR to chemotherapy and antibiotics. By contrast, many SLC family proteins [e.g., organic cation transporters, organic anion transporters, and peptide transporters (PEPTs)] may act as uptake transporters. With the understanding of pharmacoepigenetics, there is increasing evidence to support a critical role for miRNAs in posttranscriptional regulation of efflux and uptake transporter genes (Table 2), which may subsequently modulate intracellular drug accumulation and drug response.
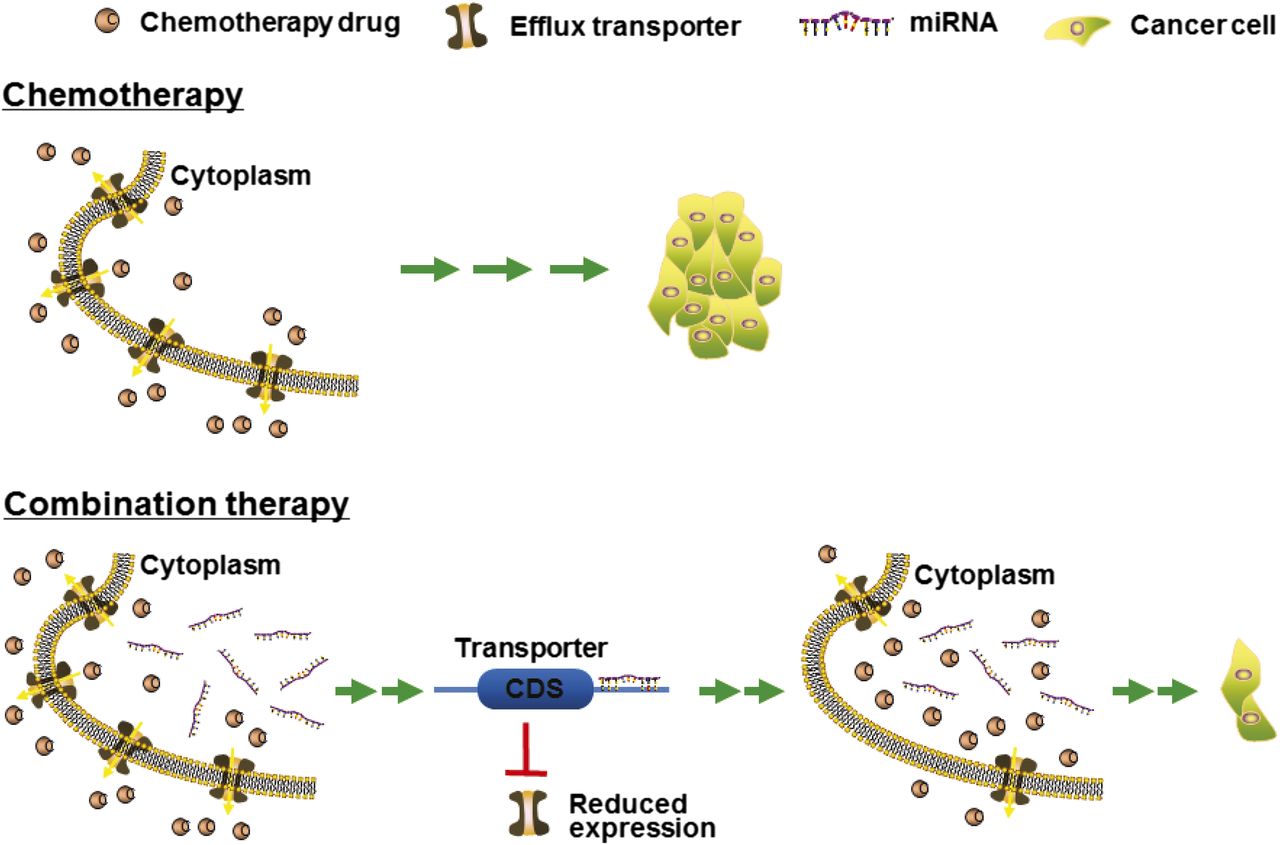
P-gp/MDR1/ABCB1 was first shown to be regulated by miR-27a and miR-451 (Kovalchuk et al., 2008; Zhu et al., 2008). Compared with drug-sensitive parental cell lines, there was an upregulation of ABCB1 as well as dysregulation of a number of miRNAs including miR-451 within drug-resistant cells. The actions of miR-451 in the regulation of ABCB1 were then demonstrated by computational analysis, luciferase reporter assay, and Western blot studies. Furthermore, treatment with miR-451 sensitized the ABCB1-overexpressing breast cancer cells to doxorubicin, suggesting that miRNAs may be employed to improve chemotherapy or combat MDR. Later, the effectiveness of combined use of miR-451 and an existing anticancer drug (e.g., irinotecan) in treating stem-like colon cancer cells was also demonstrated, which also involved targeting of the ABCB1 efflux transporter (Bitarte et al., 2011). However, some studies (Zhu et al., 2008; Li et al., 2010c; Zhang et al., 2010) showed an upregulation of ABCB1 by miR-27a, whereas others (Chen et al., 2013) reported a downregulation. Among documented ABCB1 regulatory miRNAs (Table 2), miR-508-5p was shown to reverse MDR not only in gastric cancer cells in vitro but also in xenograft tumor mouse models in vivo (Shang et al., 2014), supporting the strategy to develop miRNA-based combination therapy (Fig. 3).

Coadministration of miRNAs with chemotherapeutic agents may be more effective to control tumor progression. There is growing evidence supporting the feasibility of combination therapy. MicroRNAs downregulate the expression of efflux transporters in tumor cells, resulting in a higher level of intracellular drug accumulation and thus an improved efficacy (Zhu et al., 2008; Pan et al., 2009g, 2013; Liang et al., 2010; Shang et al., 2014; Li et al., 2015). In addition, miRNAs may directly target (proto-)oncogenes and thus additively or synergistically suppress tumor growth (Deng et al., 2014; Zhao et al., 2015).
BCRP/ABCG2, another important efflux transporter in ADME and MDR, is also regulated by a number of miRNAs via direct or indirect mechanisms (Table 2). The MDR of S1 colon carcinoma cell line is attributable to the overexpression of ABCG2. miR-519c was found to suppress ABCG2 expression in drug-sensitive parental S1 colon cancer cells via targeting of a distal miR-519c MRE site, whereas drug-resistant S1 colon cancer cells were found to consist of truncated ABCG2 transcripts lacking the distal miR-519c MRE site; together this provides a mechanistic understanding of ABCG2-mediated MDR in these cells (To et al., 2008, 2009). miR-328 was also revealed to regulate ABCG2 and consequently improve the chemosensitivity of ABCG2-overexpressing drug-resistant cells (Pan et al., 2009g). Another proximal MRE site for miR-519c (and miR-520h) were further identified and validated through luciferase report assay and mutagenesis (Li et al., 2011). In addition, the efficiency of miR-328, -519c, and -520h in the regulation of ABCG2 in human breast cancer cell lines was compared side by side (Li et al., 2011). Studies using gain- and loss-of-miRNA function approaches revealed that miR-519c and miR-328 were more effective than miR-520h in the modulation of ABCG2 protein outcome in MCF-7 cells (Li et al., 2011). As a result, both miR-519c and -328 had significant impact on intracellular accumulation of mitoxantrone, an ABCG2 substrate (Li et al., 2011). Therefore, understanding miRNA-controlled regulation not only advances the role of miRNAs in MDR but also offers clue to develop more effective therapeutic strategy, e.g., to use miRNAs to improve chemosensitivity (Fig. 3).
Many MRP/ABCC transporters are regulated by particular miRNAs (Table 2). Compared with wild-type MCF-7 cells, overexpression of ABCC1 in drug-resistant MCF-7 cells was associated with an altered miRNA expression profile, in which miR-326 was downregulated over 12-fold (Liang et al., 2010). Further studies demonstrated that miR-326 directly acted on the 3′UTR of ABCC1, reduced ABCC1 mRNA and protein expression, and sensitized drug-resistant MCF-7 cells to chemotherapy (Liang et al., 2010). Other ABCC1 regulatory miRNAs evaluated by biologic experiments include miR-134 and miR-7 in small cell lung cancer cells (Guo et al., 2010; Liu et al., 2015), miR-1291 in pancreatic cancer cells (Pan et al., 2013), and miR-133a in hepatocarcinoma cells (Ma et al., 2015). Likewise, restoring the expression or function of such ABCC1 regulatory miRNAs increased intracellular drug accumulation and chemosensitivity, supporting the feasibility of miRNA-based strategies to improve therapy (Fig. 3). In addition, some other miRNAs might modulate the expression of ABCC1 and other transporters and be implicated in the progression of hepatocarcinoma tissues (Borel et al., 2012).
The reduction of ABCC2 protein expression by rifampicin in HepG2 cells was associated with an increased ABCC2 mRNA level and altered miRNA expression profile (Haenisch et al., 2011). Among these altered miRNAs, miR-379 was elevated and might directly target ABCC2 mRNA. Indeed pre-miR-379 showed a dose-dependent suppression of ABCC2 protein expression in HepG2 cells, and miR-379 antagomir was able to rescue rifampicin-reduced ABCC2 protein expression, demonstrating a mechanistic role for miR-379 in rifampicin-altered ABCC2 protein expression at the posttranscriptional level (Haenisch et al., 2011). Another study showed that miR-297 was able to modulate ABCC2 expression in drug-resistant HCT116 cells and thus increased the efficacy of anticancer drugs (Xu et al., 2012a).
By using multiple approaches, a very recent study demonstrated that miR-124a and miR-506 was effective in modulating the expression of ABCC4 in HEK293T/17 cells, likely through a translation inhibition mechanism (Markova and Kroetz, 2014). The reduction of ABCC4 protein outcome by miR-124a and miR-506 was translated into a significantly lower level of efflux of ABCC4 substrate drug, bimane-glutathione. Although there are six major ABCC4 3′UTR haplotypes at a frequency greater than 5% among Caucasians, African Americans, and Asians, none of the inferred polymorphisms were located within or in proximity to the MRE sites for miR-124a and miR-506 and thus showed any significant effects on miR-124a- and miR-506-mediated regulation of ABCC4. In addition, miR-124a and miR-506 levels were found to be inversely correlated with ABCC4 levels in 26 human kidney samples (Markova and Kroetz, 2014), illustrating the clinical significance of miRNA-controlled regulation of ABC transporters.
There are also some reports on posttranscriptional gene regulation of SLC transporters (Table 2). The increase of human peptide transporter 1 (PEPT1/SLC15A1) protein expression during differentiation of intestinal Caco2-BBE cells was associated with three- to four-fold decrease of mature miR-92b levels (Dalmasso et al., 2011). miR-92b was then shown to directly target the 3′UTR of SLC15A1, reduce SLC15A1 mRNA and protein expression, and subsequently inhibit cellular uptake of a tripeptide (lysine-proline-valine) substrate and suppress bacterial peptide-induced proinflammatory responses in cells (Dalmasso et al., 2011). Another uptake transporter, monocarboxylate transporter 1, was also found to be posttranscriptionally regulated by several miRNAs including miR-124, miR-29a, and miR-29b (Li et al., 2009; Pullen et al., 2011).
MicroRNAs Regulate the Expression of Xenobiotic Receptors or Transcription Factors to Modulate Drug Metabolism and Disposition.
Transcriptional regulation of drug-metabolizing enzyme and transporter gene expression is primarily controlled by xenobiotic receptors or transcription factors [e.g., aryl hydrocarbon receptor (AHR), PXR/NR1I2, CAR/NR1I4, peroxisome proliferator-activated receptor α (PPARα or NR1C1), /NR1I1, nuclear factor (erythroid-derived 2)-like 2 (Nrf2 or NFE2L2), estrogen receptor alpha (ERα or NR3A1), and 9-cis retinoic acid receptor alpha (RXRα or NR2B1), etc.]. Upon activation by corresponding ligands, xenobiotic receptors and transcription factors bind to target promoter elements and lead to the upregulation of drug-metabolizing enzymes (e.g., CYP3A4 and UGT1A1) and transporters (e.g., ABCB1 and ABCG2). Therefore, miRNAs may alter the expression of enzymes and transporters through posttranscriptional regulation of xenobiotic receptor or transcription factor genes (Table 3) and thus modulate drug disposition and response (Fig. 1).
The actions of miR-27b/a illustrate miRNA-NR-enzyme/transporter signaling in the modulation of cellular drug metabolism and disposition. In addition to direct targeting of CYP3A4 (Pan et al., 2009a), CYP1B1 (Tsuchiya et al., 2006), and ABCB1 (Zhu et al., 2008), miR-27b/a was able to act on a number of xenobiotic receptors and transcription factors including NR1I1/ (Pan et al., 2009a), NR1C1/PPARα (Kida et al., 2011), ERα/NR3A1 (Li et al., 2010a), NFE2L2/Nrf2 (Narasimhan et al., 2012), and RXRα/NR2B1 (Ji et al., 2009). Therefore, alteration of enzyme/transporter protein outcome by miR-27b/a could be an accumulative effect from direct targeting of the enzyme/transporter and “indirect” targeting of regulatory factors including xenobiotic receptors and transcription factors (Yu, 2009; Yu and Pan, 2012). It should be noted that the miRNA-NR-enzyme/transporter signaling inevitably affects the mRNA levels of an enzyme/transporter, which might complicate the interpretation of miRNA regulatory mechanisms if overlooked.
PXR/NR1I2 controls transcriptional expression of many important ADME genes including CYP3A4, UGT1A, and ABCB1. miR-148a was shown to target NR1I2 and then influence CYP3A4 expression (Takagi et al., 2008). The miR-148a MRE site within NR1I2 3′UTR was defined by luciferase reporter assay. Further studies demonstrated that miR-148a precursor and antagomir were able to decrease and elevate, respectively, NR1I2 protein levels in HepG2 cells. In addition, transfection of LS-180 cells with miR-148a precursor attenuated NR1I2-dependent induction of CYP3A4 mRNA expression, suggesting a miR-148a-NR1I2-CYP3A4 axis.
CAR/NR1I3 is another important xenobiotic receptor that transcriptionally regulates the expression of various enzymes (e.g., CYP2B6 and UGT1A1) and transporters (e.g., ABCB1 and ABCC2). NR1I3 was found to be directly targeted by miR-137, which not only suppressed the protein expression of ABCB1 and CYP2B6 in drug-resistant cells but also sensitized the cells to doxorubicin in vitro as well as neuroblastoma xenografts to doxorubicin in vivo (Takwi et al., 2014). A separate study also demonstrated the actions of miR-137 in the regulation of murine Car/Nr1i3 (Chen et al., 2014a). Although miR-137 and mouse Nr1i3 expression levels were inversely related in mice during development, knocking down miR-137 led to higher rate of bilirubin clearance, validating the miR-137-Nr1i3-Ugt1a1 link in a whole body system. In addition, ectopic expression of miR-137 suppressed Nr1i3 protein levels in primary mouse hepatocytes, which could be attenuated by miR-137 antagomir.
Upon ligand binding, transcription factor AHR activates the expression of many enzymes including CYP1A1, CYP1A2, and CYP1B1. There is also evidence suggesting that CYP enzymes might be regulated by miRNA through AHR pathways (Table 3). For instance, in vivo administration of locked nucleic acid-modified antisense miR-29 antagomir to mice altered the expression of many hepatic proteins, including the upregulation of Ahr, Cyp1a1, Cyp2b10, Cyp3a11, and Cyp4a12 (Kurtz et al., 2015). A conserved miR-29 MRE site within the 3′UTR of human AHR and mouse Ahr was then validated by luciferase reporter assay, demonstrating that AHR is a direct target of miR-29. Another study showed that AHR was modulated by miR-124 in neuroblastoma SK-N-SH cells (Huang et al., 2011). In addition, miR-24 was shown to regulate the AHR dimer partner, aryl hydrocarbon receptor nuclear translocator, and thus interfere with expression and induction of CYP1A1 in Huh-7 and HepG2 cells (Oda et al., 2012).
Hepatocyte nuclear factor 4α (HNF4α/NR2A1) is an important nuclear transcription factor that binds to target DNA segment as a homodimer and serves as a master regulator of ADME gene expression. For instance, HNF4α is essential for PXR- and CAR-mediated transcriptional activation of CYP3A4 (Tirona et al., 2003). Some miRNAs were shown to directly regulate HNF4α expression and thus affect cellular ADME capacity (Table 3). Both endogenous and exogenous HNF4α protein outcome was reduced by miR-24 and miR-34a in cell line models, which likely acted through direct targeting of corresponding MRE sites (Takagi et al., 2010). As a result, introduction of miR-24 and miR-34a into HepG2 cells was able to suppress the mRNA levels of several HNF4a target genes, including CYP7A1, CYP8B1, and CYP27A1, which was reasoned to affect bile acid homeostasis (Takagi et al., 2010).
PPARα/NR1C1 is a major transcription factor in the control of lipid metabolism, which may be posttranscriptionally regulated by a number of miRNAs (Table 3). Both miR-21 and miR-27b were shown to modulate PPARα protein expression levels but not mRNA levels in Huh7 cells (Kida et al., 2011). The miR-21 MRE site within PPARα 3′UTR was further assessed by luciferase reporter assay, and the significance of miR-21 in the control of PPARα protein expression was manifested by the inverse correlation between miR-21 and PPARα protein levels in human liver samples (Kida et al., 2011). In addition, miR-506 was revealed to directly act on the 3′UTR of PPARα and modulate the protein levels of PPARα in cells (Tong et al., 2011). Although miR-506 levels were sharply higher in hydroxycamptothecin (HCPT)-resistant human colon cancer SW1116/HCPT cells, miR-506 antagomir was effective to sensitize the cells to HCPT (Tong et al., 2011), supporting the development of miRNA-based strategy to combat MDR and improve cancer therapy (Fig. 3).
The redox-sensitive transcription factor Nrf2 plays a pivotal role in cellular response to oxidative and electrophilic stress via the induction of many drug-metabolizing enzymes [e.g., NAD(P)H:quinone oxidoreductase 1 and 2 (NQO1 and NQO2), glucose-6-phosphate dehydrogenase, GSTs, and UGTs] and efflux transporters (e.g., ABCG2, ABCC3, and ABCC4). Although inherently cytoprotective, recent evidence has demonstrated that Nrf2 overexpression may actually impart MDR by regulation of xenobiotic disposition in certain cancer cells (for review, see Leinonen et al., 2014). Additionally, evidence for miRNA crosstalk within Nrf2 signaling pathway has been reported (Table 3). Among them, miR-144 was demonstrated to act directly on Nrf2 3′UTR and then modulate the expression of Nrf2 downstream targets and cellular glutathione levels (Sangokoya et al., 2010). Another study (Shi et al., 2014a) revealed that, compared with parental HepG2 cells, miR-340 was downregulated and Nrf2 was upregulated in cisplatin (CDDP)-resistant HepG2/CDDP cells. The effects of miR-340 mimics on Nrf2 and target gene (e.g., NQO1) expression were also demonstrated, which could be attenuated by miR-340 antagomir. Furthermore, administration of miR-340 mimics improved the sensitivity of HepG2/CDDP cells to cisplatin, whereas miR-340 antagomir could restore drug resistance, suggesting an important role for miR-340 in the development of cisplatin resistance in HepG2/CDDP cells and supporting the use of miRNA-based strategy to combat MDR (Fig. 3).
Conclusions
The focus on miRNA pharmacoepigenetics and pharmacoepigenomics is rising because a growing number of studies demonstrate the roles of noncoding miRNAs in the control of drug disposition and response. Although one miRNA (e.g., miR-27b) can regulate various ADME genes (e.g., CYP3A4) via direct and/or indirect targeting, the same ADME gene (e.g., ABCG2) may be modulated by multiple miRNAs (e.g., miR-328, miR-519c, and miR-520h) (for review, see Yu and Pan, 2012). As a result, miRNA signaling may have significant effects on ADME processes and drug response. Nevertheless, current understandings are mainly obtained from cell model systems and ex vivo tissue samples. Better physiologic models and in vivo studies are highly warranted to provide more direct evidence to support the importance of miRNAs in the regulation of pharmacokinetics and dynamics.
The knowledge of miRNA pharmacoepigenetics/epigenomics not only provides novel insights into interindividual variability in drug disposition and response (Yu, 2009; Yu and Pan, 2012; Ingelman-Sundberg et al., 2013; Zhong and Leeder, 2013) but also offers new clues to developing more effective treatments (Fig. 3). MicroRNAs are able to suppress the expression of some efflux transporters and enzymes and lead to a greater degree of intracellular drug accumulation, resulting in higher efficacy (Zhu et al., 2008; Pan et al., 2009g; Liang et al., 2010; Pan et al., 2013; Shang et al., 2014; Li et al., 2015). Meanwhile, some miRNAs may directly reduce the protein outcome of pharmacological targets [e.g., (proto)oncogenes] and thus control disease progression (e.g., tumor growth and metastasis) (Deng et al., 2014; Zhao et al., 2015). However, research on miRNA pharmacoepigenetics and development of miRNA-based therapy may be limited by the utilization of synthetic miRNA agents consisting of excessive artificial modifications on the phosphate linkages and/or ribose rings and thus exhibit different physicochemical and biologic properties or toxicities. By contrast, recombinant RNA technology has been successfully employed to cost-effectively produce large quantities of miRNA agents (Li et al., 2014b; Chen et al., 2015; Li et al., 2015; Wang et al., 2015), which are comprised of no or posttranscriptional modifications on the nucleobases and may better capture the structures, functions, and safety properties of natural RNAs. These natural ncRNA agents are produced in cells and should represent a novel class of miRNA agents for research and development.
Authorship Contributions
Performed data analysis: Yu, Tian, Tu, Ho, and Jilek.
Wrote or contributed to the writing of the manuscript: Yu, Tian, Tu, Ho, and Jilek.
Footnotes
- Received September 30, 2015.
- Accepted November 12, 2015.
A.-M. Yu was supported by the National Institutes of Health National Cancer Institute [Grant 1U01CA17531].
Abbreviations
- ABC
- ATP-binding cassette
- ADME
- absorption, distribution, metabolism, and excretion
- AHR
- aryl hydrocarbon receptor
- BCRP or ABCG2
- breast cancer resistance protein
- CAR or NR1I3
- constitutive androstane receptor
- CDDP
- cisplatin
- CYP or P450
- cytochrome P450
- DGCR8
- DiGeorge syndrome critical region 8
- ERα or NR3A1
- estrogen receptor alpha
- GST
- glutathione S-transferase
- HCPT
- hydroxycamptothecin
- HNF4α or NR2A1
- hepatocyte nuclear factor 4 alpha
- MDR
- multidrug resistance
- miR or miRNA
- microRNA
- miRISC
- miRNA-induced silencing complex
- MRE
- miRNA response element
- MRP or ABCC
- multidrug resistance-associated protein
- ncRNA
- noncoding RNA
- NQO
- NAD(P)H:quinone oxidoreductase
- NR
- nuclear receptor
- Nrf2 or NFE2L2
- nuclear factor (erythroid-derived 2)-like 2
- PEPT
- peptide transporter
- P-gp or ABCB1 or MDR1
- P-glycoprotein
- PPARα or NR1C1
- peroxisome proliferator-activated receptor alpha
- pre-miRNA
- precursor miRNA
- pri-miRNA
- primary miRNA
- PXR or NR1I2
- pregnane X receptor
- RISC
- miRNA-induced silencing complex
- RXRα or NR2B1
- 9-cis retinoic acid receptor alpha
- SLC
- solute carrier
- SULT
- sulfotransferase
- TRBP
- transactivation-response RNA-binding protein
- UGT
- uridine 5′-diphospho-glucuronosyltransferase
- 3′UTR
- 3′-untranslated region
- NR1I1
- vitamin D receptor
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}