


Abstract
Benzbromarone (BBR) is a benzofuran derivative that has been quite useful for the treatment of gout; however, it was withdrawn from European markets in 2003 because of reported serious incidents of drug-induced liver injury. BBR-induced hepatotoxicity has been suggested to be associated with the formation of a quinone intermediate. The present study reported epoxide-derived intermediate(s) of BBR. An N-acetylcysteine (NAC) conjugate derived from epoxide metabolite(s) was detected in both microsomal incubations of BBR and urine samples of mice treated with BBR. The NAC conjugate was identified as 6-NAC BBR. Ketoconazole suppressed the bioactivation of BBR to the epoxide intermediate(s), and the CYP3A subfamily was the primary enzyme responsible for the formation of the epoxide(s). The present study provided new information on metabolic activation of BBR.
Introduction
Gout is a form of inflammatory arthritis caused by elevation of blood urate levels (a condition known as hyperuricemia), which crystallize and deposit into the joints and/or surrounding tissues (Saag and Choi, 2006; Azevedo et al., 2014). Benzbromarone (BBR) is a benzofuran derivative (as shown in Fig. 1) that acts as a uricosuric agent by inhibiting urate reabsorption (Shin et al., 2011). Both BBR and 6-hydroxy BBR (a metabolite of BBR) have been reported to show potent human uric acid transporter 1 inhibition property (Wempe et al., 2011), which has made BBR a quite useful antigout agent for approximately 30 years in many countries. More recently, however, clinical cases of acute liver damage, including some fatalities related to BBR (Wagayama et al., 2000; Arai et al., 2002; Reinders et al., 2007), have drawn our attention to the metabolism profiles of BBR.

The chemical structure of BBR.
Hepatotoxicity is often associated with metabolic activation mediated by cytochromes P450 (P450) (He et al., 2015). Debromination was initially considered a main bioactivator of BBR in vivo (Broekhuysen et al., 1972; Ferber et al., 1981). It was clarified in 1988 that hydroxylation rather than debromination was the predominant metabolic pathway of BBR (Walter-Sack et al., 1988). Early metabolic studies revealed two major hydroxylated metabolites identified as 1′-hydroxy BBR and 6-hydroxy BBR (De Vries et al., 1993; Walter-Sack et al., 1998). It was also reported that P450s 2C9 (major) and 2C19 (minor) were involved in the formation of the 6-hydroxy BBR metabolite (De Vries et al., 1993), characterized by comparison with synthetic standard (McDonald and Rettie, 2007). Initially, BBR was found to be a P450 2C9 inhibitor (Marques-Soares et al., 2003; Locuson et al., 2004). Subsequently, BBR and four of its analogs were discovered to exhibit extraordinary inhibitory potency for P450 2C19 (Locuson et al., 2004). Idiosyncratic hepatotoxicity of BBR has primarily been suggested to be associated with metabolite 6-hydroxy BBR proposed by McDonald and Rettie (2007), who suggested that sequential oxidation of 6-hydroxy BBR results in a catechol structure, 5,6-dihydroxy BBR, which can be further oxidized to a reactive quinone intermediate capable of adducting protein; however, we believe the initial epoxidation of BBR may be more important for metabolic activation of BBR. Epoxide-derived metabolites of many protoxicants, such as bromobenzene (Slaughter and Hanzlik, 1991; Zheng and Hanzlik, 1992a), naphthalene (Zheng et al., 1997; Morisseau et al., 2008), styrene (Carlson GP, 2011), and coumarin (Born et al., 2000) are suggested to play important roles in the development of toxicities.
The objectives of this study included: 1) characterization of epoxide-derived metabolite(s) of BBR in vitro and in vivo and 2) identification of cytochrome P450 enzymes responsible for the formation of the metabolite(s). We anticipated that the study would allow us to understand better the mechanisms of idiosyncratic hepatotoxicity of BBR.
Materials and Methods
Chemicals and Materials.
BBR (purity > 98%) was obtained from Aladdin Industrial Technology Co., Ltd. (Shanghai, China). N-acetylcysteine (NAC), ketoconazole, and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from Sigma-Aldrich Co. (St. Louis, MO). Recombinant human P450 enzymes were purchased from BD Gentest (Woburn, MA). Dexamethasone (DEX), sodium nitrite, and tin chloride were purchased from the National Institute for the Control of Pharmaceutical and Biologic Products (Shenyang, China). All organic solvents were from Fisher Scientific (Springfield, NJ). All reagents and solvents were of either analytical or high-performance liquid chromatography (HPLC) grade.
Animal Experiments.
Male Kunming mice (20 ± 2 g) were obtained from the Animal Center of Shenyang Pharmaceutical University. The animals were maintained on standard mouse chow and tap water ad libitum in a 25°C room with a 12-hour dark/light cycle. Mice were individually placed in metabolism cages. After fasting for 12 hours with free access to water before the experiment, mice were treated i.p. with BBR dissolved in corn oil (10 ml/kg) at 65 mg/kg. Urine samples were collected from 0–24 hours after dosing. Control animals treated with corn oil were included. During the experiment, the animals were allowed free access to food and water. Individual groups (BBR-treated and controls) contained four mice. The collected urine samples were stored at −20°C until analysis.
Sample Preparation.
The collected urine samples were pooled and mixed with triple volumes of acetonitrile (ACN). All samples were vortexed for 3 minutes and then centrifuged at 16,000 rpm for 10 minutes at 4°C. The supernatants were harvested, and the ACN was evaporated under a stream of nitrogen gas at 40°C. The resulting urine samples were extracted with ethyl acetate (Wu et al., 2012). The organic layer was collected and evaporated to dryness under a stream of nitrogen gas at 40°C. The residues were reconstituted with 100 μl of 50% ACN in water. After centrifugation, the supernatants (5 μl) were injected onto liquid chromatography-tandem microscopy (LC-MS/MS) for analysis.
Microsomal Incubations.
Mouse liver microsomes (MLMs) were prepared in our laboratory according to previous published methods (Lin et al., 2007). DEX-induced mouse liver microsomes (MLMs) were prepared from mice pretreated with DEX (an inducer of CYP3A4) for 5 consecutive days using the same procedure. A stock solution of BBR was prepared in methanol. The incubation mixtures were prepared in a final volume of 0.5 ml of phosphate buffer (pH 7.4) containing MLMs or DEX-induced MLMs (1.0 mg of protein/ml), 3.2 mM MgCl2, 75 μM BBR, and 40 mM NAC. Total content of organic solvent was maintained at <3%. The incubation reactions were initiated by the addition of NADPH (1.0 mM). Control samples containing no NADPH were included. After 60 minutes’ incubation at 37°C, the reactions were quenched by adding equal volumes of ice-cold ACN. The reaction mixtures were vortex-mixed and centrifuged at 16,000 rpm for 10 minutes to remove precipitated protein. The resulting supernatants were evaporated to dryness under a stream of nitrogen gas at 40°C and then reconstituted with 100 μl of 50% ACN in water before injected onto LC-MS/MS for analysis. Each incubation was performed in duplicate.
Human Recombinant P450 Incubations.
To determine the specific P450 enzymes involved in the formation of reactive metabolites of BBR, a total of eight human recombinant P450s, including P450s 1A2, 2A6, 2B6, 2C9, 2C19, 2E1, 3A4, and 3A5, were tested. Conditions were equivalent to the microsomal incubations except that microsomes were replaced by the individual human recombinant P450 enzymes (20 pmol enzyme with a total volume of 200 µl in each incubation). The experiments were performed in triplicate.
P450 3A Inhibition Study.
To examine the role of P450 3A subfamily in bioactivation of BBR, similar microsomal incubations as described above were performed except for inclusion of ketoconazole at concentrations of 1.0, 10, and 100 μM. The formation of the NAC-BBR conjugate was monitored by LC-MS/MS. The experiments were performed in triplicate.
Synthesis of 6-NAC BBR.
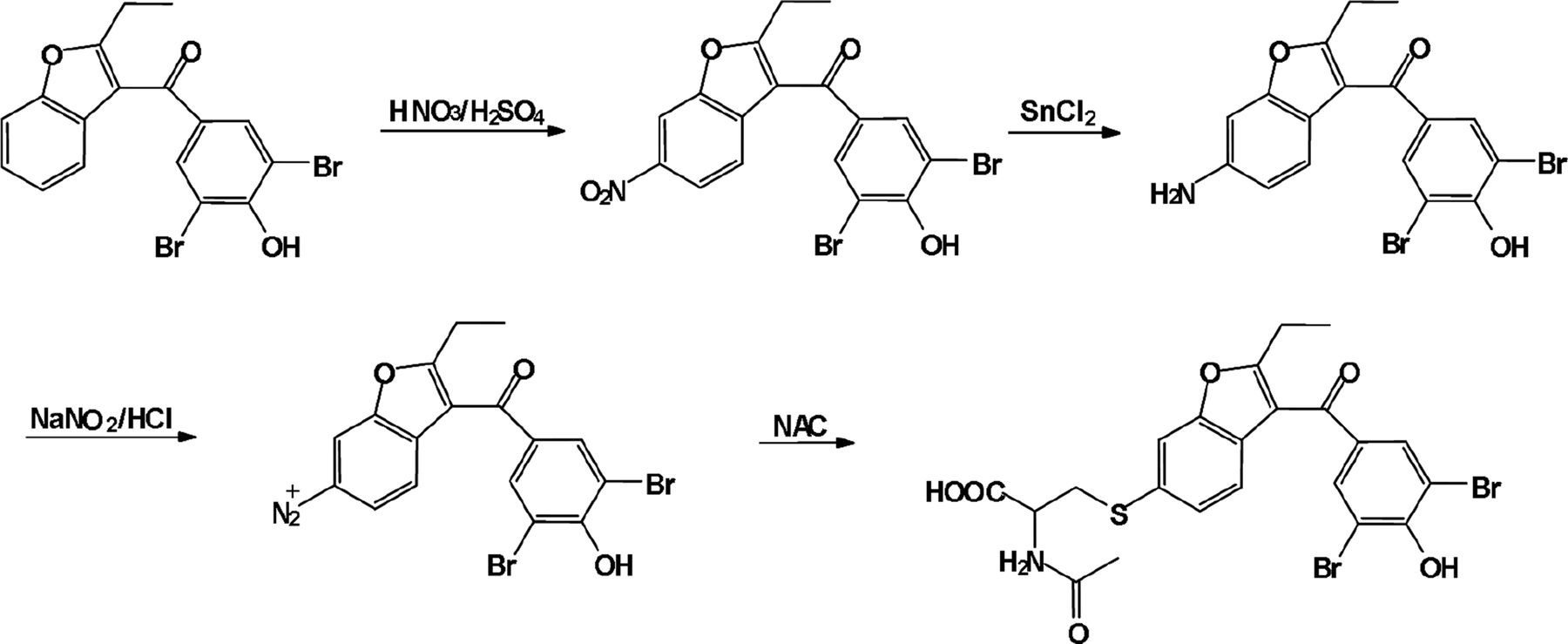
BBR (350 mg, 0.83 mmol) was slowly dissolved in 10 ml of 97% H2SO4 precooled at −15°C, followed by dropwise addition of 2 ml fuming nitric acid. The reaction mixture was constantly stirred at −15°C for 4 hours. The mixture was diluted with 20 ml of water, and yellowish solid was observed. The solid product was chromatographed over silica. The purified nitration product (220.5 mg, 0.47 mmol) was heated with SnCl2·2H2O (318.2 mg, 1.41 mmol) in 15 ml of ethanol at 80°C and refluxed for 4 hours. The mixture was neutralized to pH 7 with NaHCO3 and extracted with CH2Cl2. The remaining CH2Cl2 layer was washed with water, dried with anhydrous sodium sulfate, and evaporated. After chromatographing on silica gel, the resulting aniline (95 mg, 0.22 mmol) was dissolved in 5 ml of methanol and cooled to −10°C. To the aniline solution, two drops of concentrated hydrochloric acid added, and the mixture was diazotized by the dropwise addition of NaNO2 (22.4 mg, 0.32 mmol) in 250 μl of water. This cold diazonium solution was then added over 35 minutes to a stirred solution of NAC (105.8 mg, 0.65 mmol) in 5 ml of water held at 65°C. After the solution was stirred for 2 hours, the mixture was cooled down to room temperature and extracted with CH2C12. The CH2C12 extracts were chromatographed on silica gel (Zheng and Hanzlik, 1992b). Further purification by a semipreparative HPLC system offered 3.2 mg of BBR-NAC conjugate. All NMR measurements were obtained at 600 MHz on a BRUKER-ARX-600 spectrometer.
LC-MS/MS Method.
LC-MS/MS analyses were performed on an AB SCIEX Instruments 4000 Q-Trap (Applied Biosystems, Foster City, CA) interfaced online with an Ekspert Ultra LC100 system (Applied Biosystems). The analytical separation was achieved on an Ultimate XB-C18 column (2.1 × 100 mm, 3 μm; Welch Scientific, Inc., Shanghai, China) with a flow rate of 0.4 ml/min, and purification by semipreparative HPLC was achieved on a YMC-Pack ODS-A column (250 × 10 mm, S-5, 12 nm; YMC Co., Ltd) with a flow rate of 3 ml/min. The mobile phase consisted of solvent A (0.1% formic acid in ACN) and solvent B (0.1% formic acid in H2O). A gradient elution was applied for analytical separation: 0–2 minutes, 10% A; 2–8 minutes, 10%–70% A; 8 to 9 minutes, 70%–100% A; 9 to 10 minutes, 100% A; 10 to 11 minutes, 100%–10% A; 11–14 minutes, 10% A. An isocratic elution was used for metabolite purification by semipreparative HPLC with 45% solvent A for 45 minutes. LC-MS/MS analyses were performed on a 5-μl aliquot of samples. Turbo Ion Spray interface for electrospray ionization was operated in positive ion mode using the following conditions: ion spray voltage, 5500 V; source temperature, 650°C; curtain gas, 20 psi; ion source gas 1, 50 psi; ion source gas 2, 50 psi; declustering potential, 50 V; entrance potential, 10 V; and cell exit potential, 5 V. The information-dependent acquisition method was used to trigger the enhanced product ion (EPI) scans by analyzing multiple reaction monitoring (MRM) signals. MS analyses were conducted with ion transitions m/z 584→277, 586→279, and 588→281 for BBR-NAC conjugate derived from epoxide, m/z 616→487, 618→489, and 620→491 for BBR-NAC conjugate derived from the corresponding quinone, and m/z 260.7→116.3 for propranolol (internal standard), respectively. The collision energy was set at 45 eV with a spread of 15 eV. Data were processed using AB SCIEX Analyst software versions 1.6.0 and 1.6.1 (Applied Biosystems).
MS/MS analyses were also conducted on an Agilent 1200 Series Rapid Resolution LC system equipped with a hybrid quadrupole time-of-flight (Q-TOF) MS system (microQ-TOF; Bruker Corporation, Billerica, MA). Mobile phase A was ACN with 0.1% (v/v) formic acid, and mobile phase B was water with 0.1% (v/v) formic acid. The MS parameters were optimized as follows: endplate offset, −500 V; capillary voltage, −4500 V; nebulizer gas pressure, 0.3 bar; dry gas, high-purity nitrogen (N2); dry gas flow rate, 4.0 liters per minute; and gas temperature, 180°C. The data were analyzed by Bruker Daltonics Data Analysis 3.4 software.
Statistic Analysis.
Statistical analyses were performed by unpaired Students’s t tests, using GraphPad Prism software (GraphPad Software, La Jolla, CA). A P value of less than 0.05, 0.01 or 0.001 was considered significantly different.
Results
MS Behaviors of BBR.
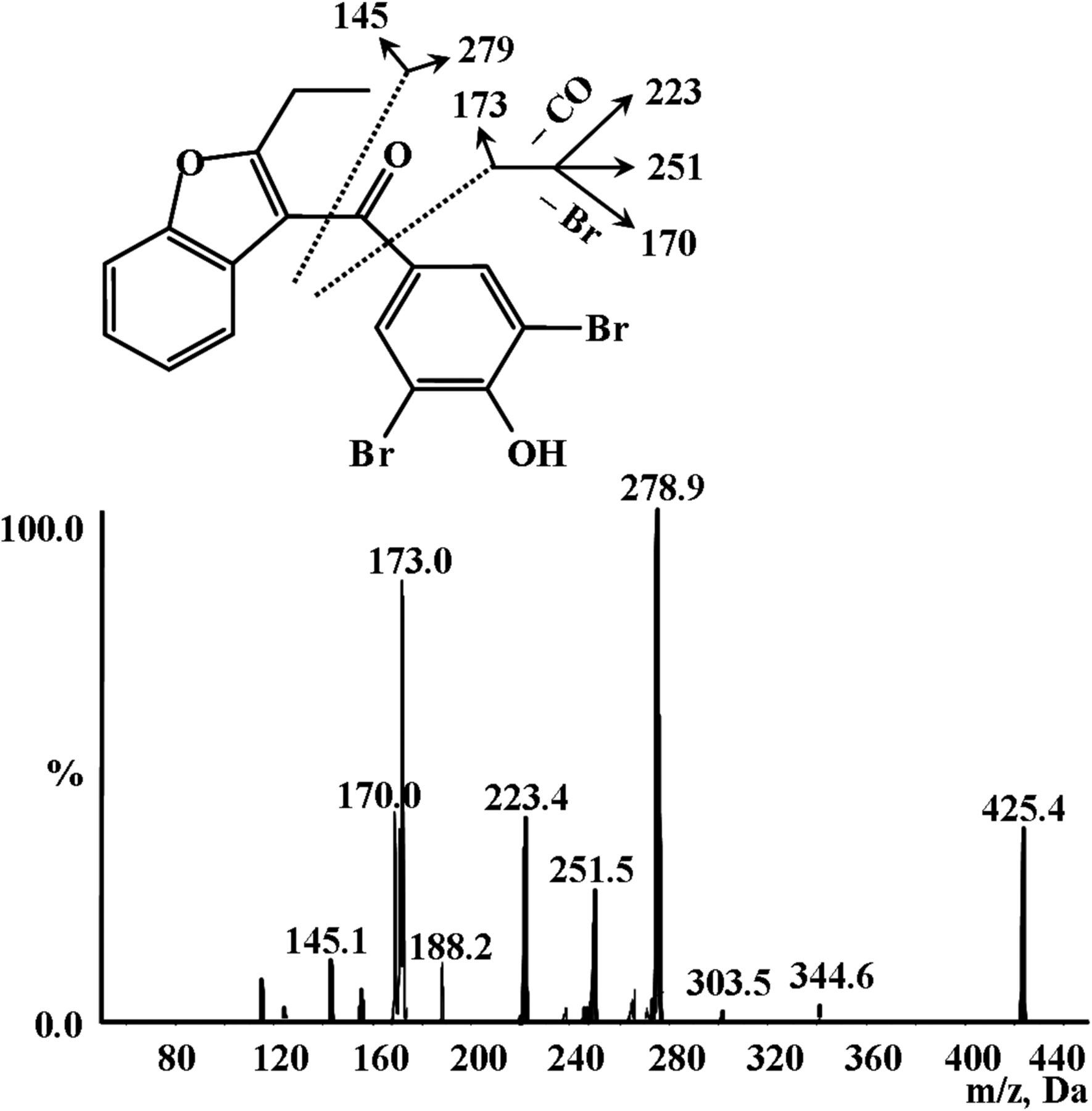
To facilitate metabolite identification, we started with MS analysis of parent compound BBR in positive ionization mode. The parent drug showed fragment ions of m/z 145, 170, 173, 223, 251, and 279 with [M + H]+ of m/z 425 (Fig. 2), which we proposed were derived from a loss of C10H9O, C6H3BrO, C11H9O2, C5H3Br2, C6H3Br2O, and C7H3Br2O2, respectively.
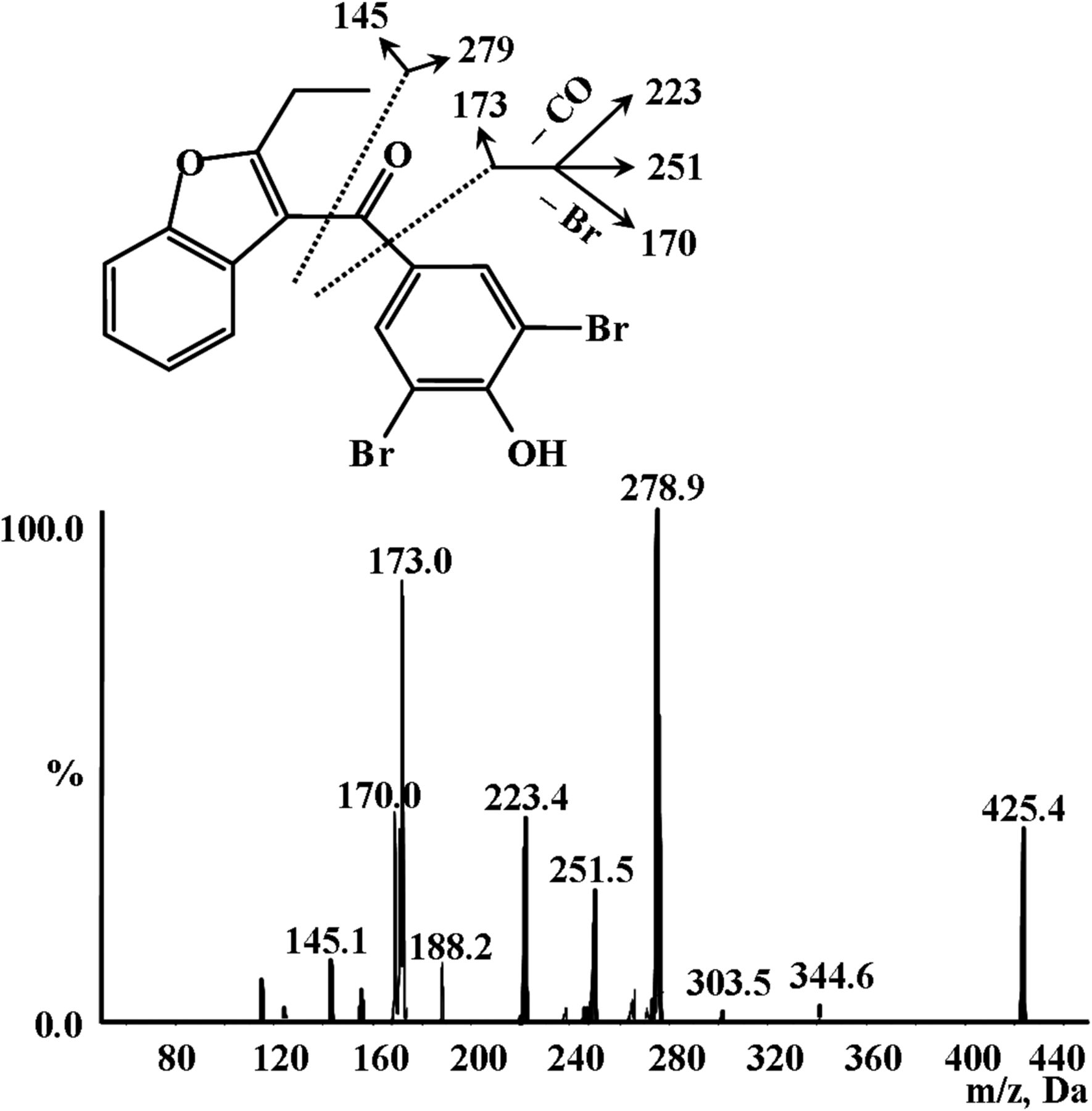
MS/MS spectrum of BBR.
In Vitro Metabolic Activation of BBR.
BBR was incubated in MLMs supplemented with NAC as a trapping agent. One metabolite (retention time = 7.99 minutes) was detected with a molecular cluster of m/z 584 (50%), 586 (100%), and 588 (50%) (Figs. 3B and 4B), suggesting an NAC incorporation in BBR. The tandem mass (MS/MS) spectrum of the metabolite obtained by MRM-EPI scanning with ion transition m/z 586→455 showed the indicative characteristic neutral loss of 129 Da associated with the cleavage of the NAC moiety (Fig. 3E). The product ions at m/z 568 and 544 were derived from the loss of H2O and acetyl group, respectively. The fragment ion at m/z 279 (loss of C7H3Br2O2) was found to be the same as that of BBR, indicating that the dibromohydroxybenzoyl ring retained unchanged. This result led us to propose that NAC was attached to the benzofuran ring. Considering that the intensity of product ion at m/z 279 was much stronger than that of m/z 455, the rest of the MS/MS spectra of the metabolite were acquired by MRM-EPI scanning with ion transition m/z 586→279 instead of m/z 586→455. The metabolite was also detected in full scan mode (Q1, Fig. 4D). No such conjugate was detected in the microsomal incubation system in the absence of NADPH (Fig. 3A), indicating that metabolism was responsible for the formation of the BBR-NAC conjugate. In addition, another metabolite with [M+H]+ of m/z 616, 618, and 620, along with retention time at 7.68 minutes, was detected in the microsomal mixture in both MRM mode (Fig. 4A) and full scan mode (Q1, Fig. 4C). The molecular ion matched the molecular weight of BBR-NAC conjugate derived from the quinone intermediate. The corresponding GSH conjugate was reported by McDonald and Rettie (2007).

Extracted ion (m/z 586→279) chromatograms obtained from LC-Q-Trap MS analysis of mouse liver microsomal incubations containing BBR and NAC in the absence (A) or presence (B) of NADPH. (C) Extracted ion (m/z 586→279) chromatogram obtained from LC-Q-Trap MS analysis of BBR-NAC conjugate generated in DEX-induced liver microsomal incubations. (D) Extracted ion (m/z 586→279) chromatogram obtained from LC-Q-Trap MS analysis of synthetic BBR-NAC conjugate. (E) MS/MS spectrum obtained from LC-Q-Trap MS analysis of BBR-NAC conjugate generated in microsomal incubation. (F) MS/MS spectrum obtained from LC-Q-Trap MS analysis of synthetic BBR-NAC conjugate. (G) MS/MS spectrum obtained from Q-TOF MS analysis of synthetic BBR-NAC conjugate.

Extracted ion chromatograms obtained from LC-Q-Trap MS analysis of BBR-NAC conjugate derived from quinone (A, m/z 618→489) and 6-NAC BBR (B, m/z 586→279) generated in microsomal incubations in MRM mode. Extracted ion chromatograms obtained from LC-Q-Trap MS analysis of BBR-NAC conjugate derived from quinone (C, m/z 618→618) and 6-NAC BBR (D, m/z 586→586) generated in microsomal incubations.
NMR Analysis of Synthetic BBR-NAC Conjugate.
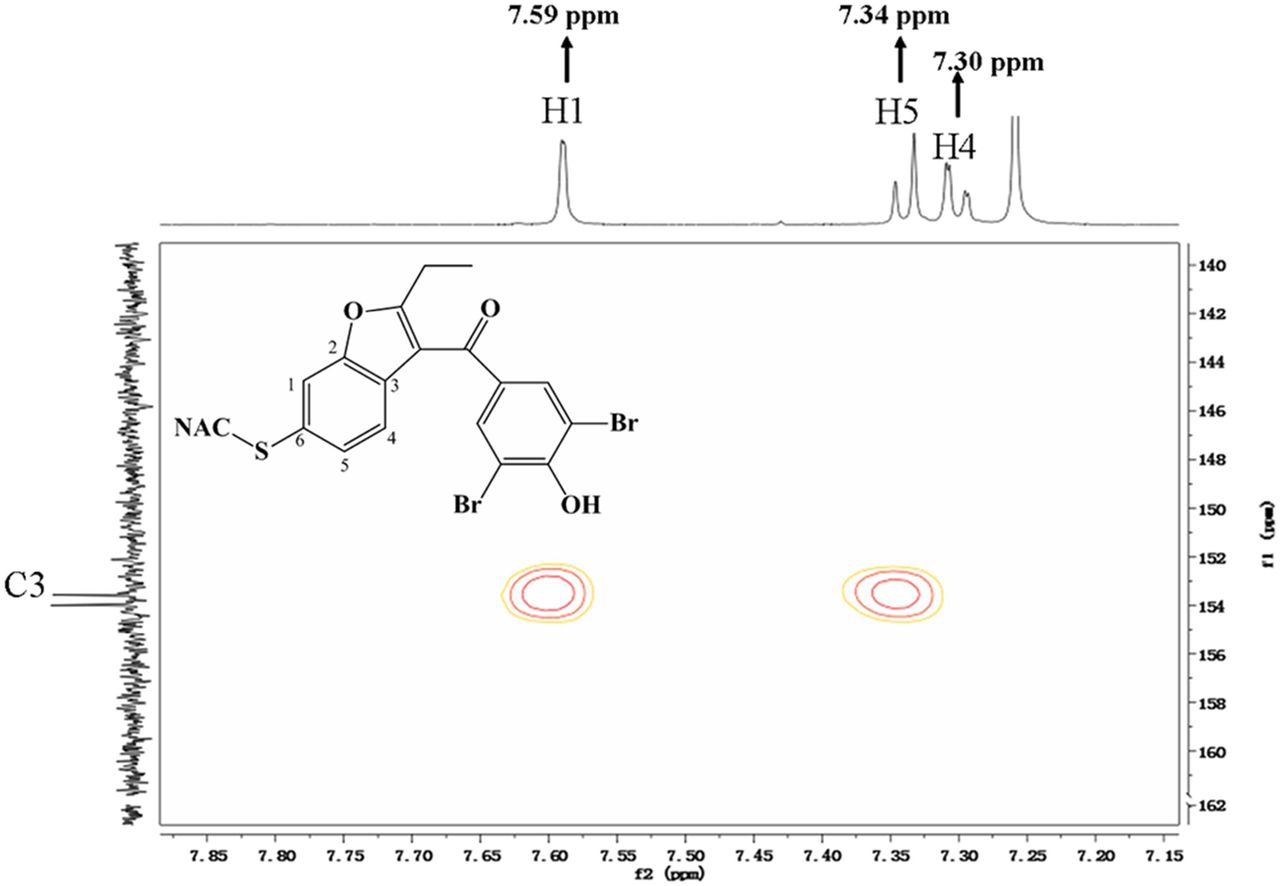
To further characterize the metabolite, we chemically synthesized the BBR-NAC conjugate (Scheme 2). Two BBR-NAC conjugates, presumably 5-NAC BBR and 6-NAC BBR, were obtained in the chemical synthesis (Supplemental Fig. 1). One product formed in the reaction showed the same chromatographic and MS identities (Fig. 3, D and F) as that for the product generated in microsomal incubations. The product was further analyzed by Q-TOF MS in positive mode. It clearly possessed the bromine isotope pattern with a molecular cluster of m/z 583.9346 (50%), 585.9353 (100%), and 587.9336 (50%) (Fig. 3G). We succeeded in obtaining 1H-NMR, 13C-NMR, and NMR spectra of the synthetic BBR-NAC conjugate. The proton NMR spectrum (Fig. 5) demonstrated three aromatic proton resonances at 7.30, 7.34, and 7.59 ppm corresponding to the protons at C4, C5, and C1 positions of 6-NAC BBR or C1, C6, and C4 positions of 5-NAC BBR. Additionally, the HMBC spectrum (Fig. 6) showed that C3 had correlations with the protons at 7.34 and 7.59 ppm, and no correlation was observed between C3 and the proton at 7.30 ppm. Thus, the identity of the conjugate was concluded to be BBR with NAC attached to C6 carbon. In other words, the conjugate was assigned to be 6-NAC BBR.

Proposed pathways for the formation of BBR-derived NAC conjugates by P450-mediated epoxidation of BBR.

Synthetic route of BBR-NAC conjugate.

1H NMR spectrum (600 MHz) of BBR-NAC conjugate.

HMBC NMR spectrum (600 MHz) of BBR-NAC conjugate.
Urinary Excretion of BBR-NAC Conjugate.
To investigate the bioactivation of BBR in vivo, the urinary excretion of BBR-NAC conjugate was monitored by a designed MRM-EPI template after an i.p. injection of BBR at 65 mg/kg in mice. As expected, 6-NAC BBR was found in the urine obtained from the animals given BBR (Fig. 7B), and no such urinary metabolite was observed before the treatment (Fig. 7A). The metabolite showed the same retention time as that for the metabolite produced in microsomal incubations (Fig. 3B).
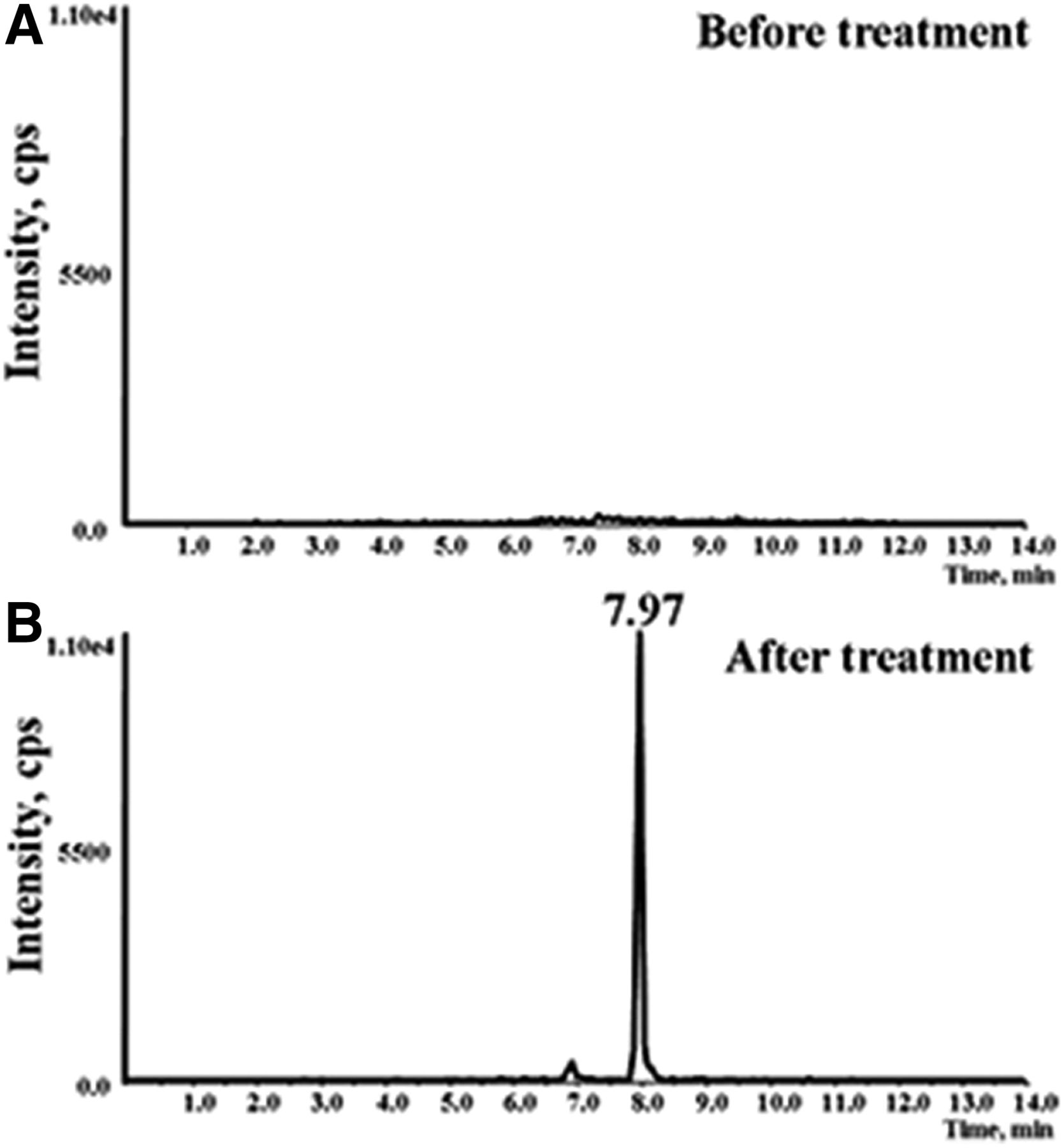
Extracted ion (m/z 586→279) chromatograms obtained from LC/Q-Trap MS analysis of urine before (A) and after (B) treatment with BBR.
Identification of P450 Enzymes Responsible for Bioactivation of BBR.
To determine which P450 enzymes preferentially catalyze the oxidation of BBR, BBR was incubated with individual human recombinant P450 enzymes. As shown in Fig. 8, 6-NAC BBR was detected in the incubations with P450 3A4. Apparently, minor or no 6-NAC BBR was observed in the incubations with the other P450 enzymes tested. The microsome inhibition studies showed that coincubation with ketoconazole reduced the production of the conjugate in a concentration-dependent manner (Fig. 9). These experiments illustrated that CYP3A subfamily was the principal enzyme responsible for the formation of epoxide metabolite(s) of BBR.

Formation of 6-NAC BBR in individual recombinant P450 enzyme incubations containing NADPH, BBR, and NAC. Data shown represent the mean ± S.D. (n = 3); *P < 0.05; **P < 0.01; ***P < 0.001 considered significantly different.

Inhibitory effect of ketoconazole at various concentrations on the formation of 6-NAC BBR in mouse liver microsomal incubations. The concentrations of ketoconazole were 1, 10, and 100 μM, respectively. Data shown represent the mean ± S.D. (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001 compared with group 3A4.
Discussion
BBR is a uricosuric agent that has been used as a treatment of chronic gout. Although it is effective, serious incidents of BBR-induced idiosyncratic hepatotoxicity including some fatalities have been reported. BBR was reported to induce impairment of oxidative phosphorylation in cultured HepG2 cells (Haegler et al., 2015). Arai et al. (2002) speculated that administration of allopurinol (xanthine oxidase enzyme inhibitor) with BBR may accelerate the occurrence of liver dysfunction. Idiosyncratic hepatotoxicity is often associated with metabolic activation mediated by cytochromes P450. A quinone-derived reactive intermediate of BBR was reported by McDonald and Rettie (2007). It was our speculation that the generation of epoxide-derived intermediate(s) would be required before the formation of the quinone-derived reactive intermediate. To seek the epoxide intermediate(s), BBR was incubated in microsomes supplemented with NAC as a trapping agent. We reasoned that the enzymatic epoxidation of BBR would give epoxides 2 and/or 3 (Scheme 1). Adduction of the epoxides with NAC would produce hydroxyl nonaromatic NAC conjugates 4–7 that may further be dehydrated to NAC conjugates 10–12. No nonaromatic NAC conjugates (4–7) were detected in the microsomal mixtures. Instead, one aromatic NAC conjugate was observed in the microsomal mixtures. This conjugate was also found in the urine samples of mice given BBR. In addition, we chemically synthesized the conjugate that displayed the same chromatographic and MS identities as that of the one observed in microsomal incubations and the urine samples. The conjugate was characterized as 6-NAC BBR (conjugate 11) by NMR. Identification of the NAC conjugate provided strong evidence for the formation of the epoxide intermediate(s).
The formation of 6-NAC BBR was NADPH-dependent, indicating that the bioactivation of BBR was mediated by cytochromes P450. The recombinant P450 enzyme studies showed that CYP3A4 was the major enzyme responsible for the generation of the arene epoxide(s) and that decreases in BBR concentrations (75, 25, and 5 μM) did not cause a large loss of the efficiency of CYP3A4 to catalyze the formation of the epoxide metabolite (Supplemental Fig. 2). Coincubation of ketoconazole suppressed the formation of the reactive metabolite(s) in microsomal reactions. Higher activity to catalyze the bioactivation of BBR was found in microsomes obtained from DEX-induced mice than that in regular microsomes (Fig. 3C). The observed potentiating effect of DEX on metabolic activation of BBR, combined with the suppressive effect of ketoconazole on the formation of the epoxide(s) in microsomal reactions with BBR, suggests the participation of CYP3A in the production of the epoxide metabolite(s) of BBR in vitro. Pretreatment with ketoconazole resulted in 60% decrease in urinary 6-NAC BBR in mice (Supplemental Fig. 3), indicating that CYP3A-mediated bioactivation of BBR took place also in vivo.
The present study provided new information on the formation of P450-mediated reactive metabolite(s) of BBR, and the metabolic process had not previously been reported. The pathway for bioactivation of BBR presumably involves epoxidation of the benzofuran ring to epoxide intermediates 2 and/or 3. Four possible hydroxyl nonaromatic NAC conjugates (4–7) would be formed after reaction with NAC. Sequential dehydration would offer three different aromatic NAC conjugates (10–12). Interestingly, only one NAC conjugate (i.e., 6-NAC BBR) was observed in microsomal incubations and urine samples. Two episulfonium ions (8 and 9, Scheme 1) as intermediates are proposed for the explanation of the observation. For some reason, the rearrangement of episulfonium ions 8 and 9 exclusively produces aromatic NAC conjugate 11.
McDonald and Rettie (2007) suggested a possible mechanism of toxicity that involves the bioactivation of BBR through sequential steps of oxidation of the benzofuran ring to a quinone intermediate, an electrophilic species reactive to nucleophilic sites of biomolecules. Although quinones are known electrophiles, epoxides 2 and 3 would be the first electrophilic metabolites generated in the line of the metabolic pathway of BBR. In the present study, the level of the epoxide-derived NAC conjugate was approximately 10-fold higher than that of the conjugate derived from the quinone in microsomes incubated with BBR (Fig. 4, C and D), and little quinone-derived NAC conjugate was detected in the urine samples of animals treated with BBR. The abundance of conjugate 11 observed in microsomal incubations and urine samples suggests that the initial epoxidation might be logically more important in metabolic activation of BBR, although further mechanistic investigation is needed.
The mechanisms underlying idiosyncratic hepatotoxicity remain largely unknown. Toxic effects of metabolites are thought to be one of the possible factors implicated (Greer et al., 2010; Xuan et al., 2015). Our work showed that CYP3A4 was the primary P450 enzyme responsible for the metabolic activation of BBR (Fig. 8). Several drugs reportedly eliciting idiosyncratic hepatotoxicities have been documented to be bioactivated mainly by CYP3A4, such as nefazodone (Kalgutkar et al., 2005), zafirlukast (Kassahun et al., 2005), rimonabant (Foster et al., 2013), and amiodarone (Takai et al., 2016; Xuan et al., 2015). CYP3A4 is the major human hepatic P450 enzyme, and the abundance of the enzyme seems unlikely to explain the rare incidence of idiosyncratic events in humans. We speculate that other factors, such as deficiency of glutathione or/and glutathione S-transferases, imbalance between cellular damage and protective responses, and oversensitivity of immune system, could also be involved in the adverse effect.
In conclusion, the present study demonstrated that microsomal incubations of BBR generated epoxide intermediate(s) that can be trapped with NAC to produce 6-NAC BBR exclusively. Ketoconazole suppressed the bioactivation of BBR to the epoxide intermediate(s), and the CYP3A subfamily was the primary enzyme responsible for the formation of the epoxide(s). The results provide additional information on bioactivation of BBR.
Authorship Contributions
Participated in research design: Zheng.
Conducted experiments: H. Wang, Peng.
Performed data analysis: K. Wang, H. Wang.
Wrote or contributed to the writing of the manuscript: Zheng, H. Wang.
Footnotes
- Received August 22, 2015.
- Accepted January 13, 2016.
K.W. and H.W. contributed equally to this work.
This work was supported in part by the National Natural Science Foundation of China [Grants 81373471 and 81430086], and the Natural Science Foundation of Liaoning Province [Grant 2015020738].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- BBR
- benzbromarone
- DEX
- dexamethasone
- EPI
- enhanced product ion
- HPLC
- high-performance liquid chromatography
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- MLM
- mouse liver microsome
- MRM
- multiple-reaction monitoring
- MS/MS
- tandem mass spectrometry
- NAC
- N-acetylcysteine
- NADPH
- β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt
- P450
- cytochrome P450
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}