


Abstract
Childhood cancer represents more than 100 rare and ultra-rare diseases, with an estimated 12,400 new cases diagnosed each year in the United States. As such, this much smaller patient population has led to pediatric oncology drug development lagging behind that for adult cancers. Developing drugs for pediatric malignancies also brings with it a number of unique trial design considerations, including flexible enrollment approaches, age-appropriate formulation, acceptable sampling schedules, and balancing the need for age-stratified dosing regimens, given the smaller patient populations. The regulatory landscape for pediatric pharmacotherapy has evolved with U.S. Food and Drug Administration (FDA) legislation such as the 2012 FDA Safety and Innovation Act. In parallel, regulatory authorities have recommended the application of physiologically based pharmacokinetic (PBPK) modeling, for example, in the recently issued FDA Strategic Plan for Accelerating the Development of Therapies for Pediatric Rare Diseases. PBPK modeling provides a quantitative and systems-based framework that allows the effects of intrinsic and extrinsic factors on drug exposure to be modeled in a mechanistic fashion. The application of PBPK modeling in drug development for pediatric cancers is relatively nascent, with several retrospective analyses of cytotoxic therapies, and latterly for targeted agents such as obatoclax and imatinib. More recently, we have employed PBPK modeling in a prospective manner to inform the first pediatric trials of pinometostat and tazemetostat in genetically defined populations (mixed lineage leukemia–rearranged and integrase interactor-1–deficient sarcomas, respectively). In this review, we evaluate the application of PBPK modeling in pediatric cancer drug development and discuss the important challenges that lie ahead in this field.
Introduction
Childhood cancer represents a collection of more than 100 rare and ultra-rare diseases, with an estimated 12,400 new cases diagnosed each year in the United States in patients aged <21 years (Ries et al., 1999). This much smaller patient population has, in large part, led to cancer drug development for pediatric patients being an afterthought, with approval for pediatric indications following the development and approval of the agent in adult cancers (Kearns et al., 2003). The rate of development has also been slow; from 1948 to 2003, there were 120 new cancer drug approvals, only 30 of which were used in pediatrics (Adamson et al., 2014). Developing drugs for pediatric malignancies also brings with it a number of unique challenges in terms of trial design, potential for disparity in genetics and pathophysiology relative to the adult disease, and what is considered acceptable clinical benefit. Trial design considerations include age-stratified dosing regimens, age-appropriate formulations (Breitkreutz and Boos, 2007), amenable sampling schemes (given the limitations on the number and volume of blood draws), as well as the necessity for flexible enrollment approaches in phase I, such as the rolling six design (Skolnik et al., 2008). The specific oncogenic pathways in adult carcinomas may not be active in the childhood malignancy, although common cellular pathways have emerged (Rossig et al., 2011). The benchmark for clinical benefit can also be quite different. Increasing overall survival by a few months may be acceptable in adults but the focus in pediatric oncology is cure, with cure rates in most cases of >70% and 5-year event free survival of approximately 80% (Adamson et al., 2014). However, novel drug approaches are needed for children with difficult-to-treat malignancies such as stage IV neuroblastoma, sarcomas, brain tumors, and relapsed leukemia (Horton and Berg, 2011). As a direct consequence of the unmet need in pediatric pharmacotherapy in general, the regulatory landscape has evolved with the introduction of U.S. Food and Drug Administration (FDA) legislation including the Best Pharmaceuticals for Children Act in 2002 and the Pediatric Research Equity Act in 2003 providing incentives for evaluating and developing drugs for children, which were both made permanent under the FDA Safety and Innovation Act in 2012.
In developing and optimizing pediatric pharmacotherapy, an appreciation of the comparative physiology and biochemistry between adults and children is paramount, being particularly acute in prenatal, infant, and toddler populations. As the long-standing adage states, “children are not small adults”; thus, simple dose reductions may not be sufficient and the nature of disease can also be divergent. As our knowledge of normal growth and development has increased, so too has our understanding of how these growth changes can greatly affect pharmacokinetics (PK) and ultimately response to therapy. Drugs in pediatric oncology are typically administered by either intravenous or oral routes; as such, there are a number of physical, chemical, and biologic barriers that must be overcome to reach the target site of action. The gastrointestinal absorption of drugs can be markedly affected by the developmental changes in absorptive surface area, intraluminal pH, splanchnic blood flow, gastric emptying time, and intestinal motility (Batchelor et al., 2014). The maturation profile in biliary function also leads to age-related differences in duodenal bile salt concentrations, which can affect drug dissolution and solubility at the site of absorption (Poley et al., 1964). The ontogeny in intestinal expression of drug-metabolizing enzymes and transporters can also be a contributing factor for oral bioavailability. This is illustrated with the alkylating antineoplastic agent, busulfan, in which biopsies of distal duodenum indicated that glutathione-S-transferase (GST) activity decreased from infancy through adolescence and manifested as a reduced oral clearance of busulfan, a GST substrate (Gibbs et al., 1999). Drug distribution can also show marked age-related changes as a result of physiologic and biochemical ontogeny. Infants and children have proportionally higher extracellular water and total body water than adults, in addition to the obvious differences in cardiac output, tissue perfusion, and organ size (Kearns et al., 2003). The major plasma proteins, albumin and α-1-acid glycoprotein (AAG), also show developmental changes, which can affect the distribution of highly protein bound drugs (Ehrnebo et al., 1971). The delayed maturation of drug-metabolizing enzymes (DMEs) and potential for toxicity was typified with the case of newborns treated with doses of the antimicrobial chloramphenicol, extrapolated from those shown to be safe and effective in adults, which led to cardiovascular collapse associated with Gray syndrome in neonates. The mechanistic basis for these drug-induced sequelae was later shown to be an immature uridine diphosphoglucuronosyl transferase (UGT) system resulting in impaired metabolic clearance. The maturation profile of DMEs, the most studied in this regard being the cytochrome P450 (P450) superfamily (Hines, 2009), can follow one of three possible trajectories: 1) the enzyme exhibits high expression during the first trimester and either remains high or decreases during gestation before a steep decline in expression level postnatally, within a few days or up to 2 years after birth (e.g., CYP3A7); 2) the enzyme shows relatively constant expression throughout gestation and postnatally (e.g., CYP2C19); and 3) the enzyme may be not expressed or present at low levels in the fetus, followed by a marked increase in expression level, depending on the isoform in question, between the second and third trimesters to the first years of life (e.g., CYP3A4). Similarly, isoforms of the other DME families can be categorized in this manner, such as the sulfotransferases, flavin monooxygenases, and UGTs. The prevalence of enzymes falling into the third class underpins the consistent observation in clinical studies of drugs metabolized by the liver showing an age-dependent increase in plasma clearance in children aged <10 years, such as dextromethorphan (Blake et al., 2007), caffeine (Pons et al., 1988), and midazolam (Altamimi et al., 2015). It should also be noted that excretory mechanisms also show an age-based relationship in terms of renal function, glomerular filtration rate, and renal blood flow that can all affect the elimination of renally excreted drugs. More detailed reviews of the physiologic and biochemical ontogeny influencing the absorption, distribution, metabolism, and excretion (ADME) of drugs were reported previously (Kearns et al., 2003; Hines, 2009).
It is clear that ontogenesis plays a key role in the metabolism and disposition of drugs in children; as a result, simplified dosing algorithms based on body size alone are usually insufficient. Traditional methods such as allometry have been used to scale pediatric doses, wherein absolute dose (or clearance) is related to the child-to-adult body weight ratio raised to the power of 3/4 (eq. 1) (Björkman, 2006; Mahmood, 2006). The basis for the 3/4 power law has been discussed elsewhere (West et al., 1999). Since this does not account for maturation processes and has a tendency to overpredict clearance in younger children, its use has been limited to children aged >1 to 2 years. Further refinement with the inclusion of terms for maturation or organ function has shown utility within predefined limits (eq. 2) but still lacks the holistic and mechanistic facets that are possible with physiologically based pharmacokinetic (PBPK) modeling. (1)
(1) (2)In eqs. 1 and 2, BW indicates body weight, CL is clearance, MF is maturation function, and OF is organ function.
(2)In eqs. 1 and 2, BW indicates body weight, CL is clearance, MF is maturation function, and OF is organ function.
PBPK modeling provides a quantitative and systems-based framework with each compartment representing a physiologic volume interconnected by flow rates that are anatomically representative of the circulatory system (Fig. 1). Mass balance equations describe the transfer of drug from the arterial blood into tissues and from tissues into venous blood. As such, simulations are dynamic in nature, with model fitting and prediction focused on time-concentration data. Importantly, PBPK models are more comprehensive than empirical PK models, incorporating both system-specific parameters as well as drug-specific parameters. Although it is not a new approach, PBPK modeling applied in pharmaceutical research and development has gained increasing attention in recent years with the availability of robust in vitro and in silico data, advancements in in vitro to in vivo extrapolation, and the substantial investment that has been made in the curated databases of system-dependent and probe drug-dependent parameters that are the foundation of the commercially available software packages. This technological progress has enhanced the number and breadth of robust applications in the area of clinical pharmacology, including first-in-human studies, special populations, drug interactions, and biopharmaceutics/formulations (Jones et al., 2015; Sager et al., 2015). Considerable experience has now accumulated with the development of PBPK models to describe drug concentration-time profiles in adults (Jones et al., 2009; Kostewicz et al., 2014), and, more recently, in cancer drug development (Block, 2015) and special populations such as pediatrics (Khalil and Läer, 2011; Barrett et al., 2012; Maharaj and Edginton, 2014). There have been multiple applications of PBPK in pediatric drug development, many of which are in support of the first pediatric trial, including starting dose selection, prediction of exposures across the age continuum, optimization of blood sampling strategy, prediction of target organ exposure [safety and pharmacodynamics (PD)], and evaluation of drug interaction potential.
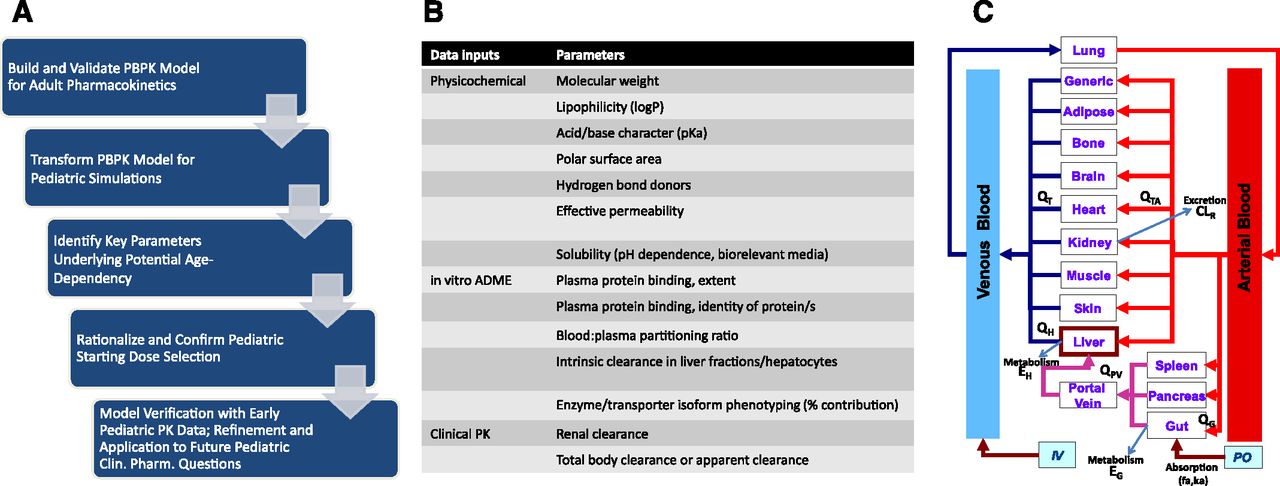
(A) A typical workflow scheme for the development of a PBPK model in support of pediatric dose selection early in clinical development. (B) Representative drug-specific data inputs used in “bottom-up” PBPK model building. (C) Generalized scheme for a PBPK model. IV, intravenous; PO, oral.
The momentum in the PBPK modeling arena has evolved to a regulatory science. As mentioned earlier, the health authorities, FDA, European Medicines Agency, and the Ministry of Health, Labor and Welfare of Japan (European Medicines Agency, 2014; Zhao et al., 2012; Shepard et al., 2015; Wagner et al., 2015) have all highlighted PBPK modeling in guidances covering drug interactions and hepatic impairment. Furthermore, PBPK modeling and simulation has recently been used to directly support labeling statements (e.g., Imbruvica; related to the use of moderate CYP3A inhibitors with ibrutinib; Imbruvica product label; Janssen, Titusville, NJ). Between 2008 and 2012, the FDA received 33 Investigational New Drug/New Drug Application submissions containing PBPK modeling approaches (including six pediatric submissions, although none in oncology). In parallel with the increasing number of submissions, the agency has increasingly used de novo (i.e., FDA initiated) PBPK modeling in its reviews to help characterize PK in a variety of complex clinical scenarios (Huang et al., 2013). In 2013, the cumulative number of submissions containing PBPK modeling increased to 84, with 22% of them related to pediatric applications (Zhao, 2014). Furthermore, the FDA recently issued a “Strategic Plan for Accelerating the Development of Therapies for Pediatric Rare Diseases,” which specifically highlights the role of modeling and simulation approaches such as PBPK modeling to inform the design and conduct of PK/PD studies and other clinical trials for investigational drugs in pediatric rare disease populations (http://www.fda.gov). Given this regulatory focus, the PBPK community is redoubling efforts to formulate industry-wide best practices and standardize the level of rigor and verification that is needed in PBPK model submissions (Sager et al., 2015).
The construction of PBPK models has typically used the various in vitro and physicochemical data available in early preclinical drug development in what is often termed a “bottom-up” approach, in an attempt to recapitulate the concentration-time data (Fig. 1). Alternatively, there may be reasons to consider a “top-down” approach in which the underlying drug-specific parameterization of the PBPK model is accomplished by optimizing the fit of the time-concentration data in question. In practice, there is usually an approach that lands somewhere between the two, termed “middle out,” in which some of the drug-specific parameters based on in vitro or in silico data may not scale appropriately or there are missing data and incomplete information regarding some aspects of drug disposition, and an element of top-down model optimization is necessary. Furthermore, mechanistic understanding can be gained through parameter sensitivity analysis and asking “what if” questions of the optimized model.
Case Studies of PBPK Modeling in Pediatric Oncology
The application of PBPK modeling in pediatric oncology drug development is still quite nascent and has largely been focused on retrospective analyses in validating the approach. A summary of reported applications is shown in Table 1. The first published model in this area of drug research dates back to 1982, with this seminal PBPK model being built with a limited number of compartments from the Bischoff–Dedrick multiorgan model. The observed parent cisplatin and total platinum serum concentrations in 14 pediatric patients were adequately recapitulated in the model and helped to better understand changes in the renal clearance of total platinum (Evans et al., 1982). Similar reports based on a modified Bischoff–Dedrick model followed and were expanded to incorporate effusion spaces as a compartment of drug distribution and clearance (Li and Gwilt, 2002). Simulated methotrexate plasma concentrations in adults without effusions were adequately modeled prior to providing congruent simulated and measured plasma and effusion methotrexate concentrations in one pediatric patient with a malignant pleural effusion after intravenous administration. This early work has been expanded on with PBPK modeling applications reported for a number of drugs used in the pediatric oncology setting, including busulfan, docetaxel, etoposide, coadministered 6-mercaptopurine and methotrexate, and, more recently, imatinib, obatoclax, pinometostat (EPZ-5676; 9-[5-deoxy-5-[[cis-3-[2-[6-(1,1-dimethylethyl)-1H-benzimidazol-2-yl]ethyl]cyclobutyl](1-methylethyl)amino]-β-d-ribofuranosyl]-9H-purin-6-amine), and tazemetostat (EPZ-6438; N-[(1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-5-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-4-methyl-4′-(4-morpholinylmethyl)-[1,1′-biphenyl]-3-carboxamide).
Summary of PBPK modeling applications in pediatric oncology drug development
Unlike most pediatric applications wherein adult PK data are leveraged in the PBPK model building process, the cisplatin model was initially developed with PK data in dogs and adjusted for human pediatric physiology (Evans et al., 1982). As summarized in Table 1, the more common approach to pediatric PBPK modeling is to scale a validated adult PBPK model to children by incorporating ontogeny and maturation processes, thereby negating the issue of potential species differences. Another exception to the adult-to-children model workflow was presented by Barrett et al. (2011). After establishing quantitative pharmacology relationships between in vitro activity data and a preclinical diseased mouse model, obatoclax exposures were recapitulated in the mouse prior to scaling to a human infant, with the goal of evaluating the potential to achieve targeted exposure in this population (Barrett et al., 2011). This approach allowed tissue distribution to be modeled and scaled, such that the PBPK model based on obatoclax physicochemical properties compared well with observed obatoclax levels in the plasma, spleen, liver, and kidney of leukemia-bearing mice for two single intravenous dose levels; by contrast, exposure values were overpredicted in the brain. After scaling to human infants, PBPK simulation of obatoclax administration to 1-year-old patients suggested that adequate drug exposure to target organs should be achievable at clinically relevant doses. To the best of our knowledge, this alternate approach for obatoclax has not been verified with clinical data from a pediatric population. By contrast, and in line with a recently proposed pediatric PBPK model development workflow (Maharaj and Edginton, 2014), Thai et al. (2015) first developed a model for docetaxel from in vitro ADME and intravenous PK data from more than 500 patients with cancer after either single or multiple dosing, to demonstrate how modeling can be applied to optimize dose and sampling times for a pediatric PK bridging study. Docetaxel is highly protein bound, has a high volume of distribution, and is highly metabolized. In the full-body PBPK model developed for adult patients, all 16 compartments were well stirred or perfusion limited, except for the liver, where organic anion transporting polypeptide 1B1 and1B3 activities were considered, and, for the muscle, where apparent passive diffusion was used. The tissue/plasma partition was determined using the volume of distribution as a primary predictor, in addition to physicochemical descriptors. Hepatic (CYP3A), biliary, and renal clearances were fit in a top-down approach. The model was customized for oncology patients by considering demographic and pathophysiological differences from healthy subjects. After scaling to a pediatric population, and as determined by visual inspection, the model adequately predicted docetaxel plasma concentration-time profiles in neonates to 18-year-old patients by accounting for age-dependent physiologic differences and CYP3A ontogeny, with predicted clearance and volume of distribution within 1.5-fold of observed data (Thai et al., 2015).
Although P450 ontogeny functions are implemented in commercial PBPK software, scaling of other metabolic pathways is less common. In that regard, the modeling exercise described by Diestelhorst et al. (2014) for the DNA-alkylating agent busulfan provides a proof of concept for GST substrates. In brief, an adult PBPK model was refined by implementing GST-A1 in 11 organs, using the PK-Sim integrated enzyme expression database, and adding irreversible DNA binding and plasma protein binding processes. Age-dependent enzyme activity and maturation factors were considered and the adult-to-child scaling indicated lower clearance values for children relative to adults. Intravenous administration of busulfan was simulated in pediatric patients, with a mean percentage error for all patients of 3.9%; 3 of 23 children demonstrated a mean percentage error of greater than ±30%, showing an adequate predictive performance of this retrospective model (Diestelhorst et al., 2014). In another study, both CYP3A4 and UGT1A1 ontogeny information was included to model etoposide exposure in children (Kersting et al., 2012). Etoposide is highly bound to plasma proteins, metabolized by CYP3A4 and UGT1A1, and eliminated in urine and bile. An adult PBPK model was developed using data from nine women with primary breast cancer receiving the drug intravenously, as part of a polychemotherapy regimen before stem cell transplantation. The physicochemical parameters of etoposide, along with the unbound plasma fraction at high and low doses in adults, kinetics for in vitro metabolic enzymes [CYP3A4 and UGT1A1], and active biliary and renal transporters [P-glycoprotein (P-gp), multidrug resistance-associated protein 2, and a hypothetical influx transporter] were incorporated into the adult model. The tissue/plasma partition coefficients were generated using the Rodgers and Rowland model. In adults, the simulated plasma concentration-time profiles of protein-bound and free etoposide were in good agreement with observed data, with mean relative deviation of 1.12 and 1.36 for low and high doses of etoposide, respectively. The model was scaled to children, incorporating ontogeny for both CYP3A4 and UGT1A1, glomerular filtration, and tubular excretion mediated by P-gp and adequately simulated the exposure seen in 18 children with various diagnoses and normal renal and liver function. Of interest, the effects of the coadministration of cyclosporine A on the metabolism and excretion of etoposide was determined in five patients, suggesting that the PBPK model could be useful for performing hypothesis testing on the effect of concomitant medications, a relatively poorly explored application of PBPK in pediatric drug development but extremely relevant given the polypharmacy in this patient population. A recent model developed for 6-mercaptopurine, a purine antimetabolite and prodrug used in combination with methotrexate as therapy for childhood acute lymphoblastic leukemia, is another example of drug–drug interaction (DDI) risk assessment by PBPK modeling in pediatric oncology (Ogungbenro et al., 2014a,b). 6-Mercaptopurine undergoes very extensive intestinal and hepatic metabolism after oral dosing, owing to the activity of xanthine oxidase leading to low and highly variable bioavailability. Dose adjustment during treatment is still based on toxicity rather than routine therapeutic drug monitoring and this work was an attempt to improve dose individualization and dosage regimen optimization through modeling and simulation, ultimately to achieve a better outcome in patients with childhood acute lymphoblastic leukemia. The PBPK model, based on the assumption of the same elimination pathways in adults and children with age-dependent parameters adjusted for pediatric scaling, adequately predicted plasma and red blood cell concentrations both in terms of population mean and variability versus observed data after intravenous or oral administration in children aged 3–18 years. Further model refinements incorporated parameters such as net secretion clearance, biliary transit time, and red blood cell distribution and binding and enabled the oral absorption of methotrexate to be well described with recapitulation of the nonlinear relationship between the fraction absorbed and the methotrexate dose. The inhibition of 6-mercaptopurine first-pass metabolism by methotrexate (via xanthine oxidase inhibition in the gut and liver) predicted an interaction but to a greater extent than that reported clinically. The predicted percentage increase in the area under the curve (AUC) and peak serum concentration (Cmax) was approximately 65% and 50% in 5- to 18-year-olds, respectively, whereas the observed increase in AUC and Cmax has been reported as 31% and 26%, respectively.
In the context of a Pediatric Investigation Plan, the European Medicines Agency (2013) requested PBPK simulations as part of the submission package for the use of imatinib (Gleevec; Novartis, Basel, Switzerland) in the treatment of pediatric patients with diagnosed Philadelphia chromosome–positive acute lymphoblastic leukemia combined with chemotherapy. PBPK modeling was used to predict area under the curve at steady state (AUCss) and concentration-time profiles in pediatric subjects, to evaluate factors influencing imatinib exposure in pediatric patients. An adult PBPK model was validated and then transformed using growth and maturation processes for pediatric scaling. The model evaluations compared the predicted versus observed imatinib AUCss and concentration-time profile from 67 pediatric patients. The simulations showed that 29 of 31 actual AUCss values normalized to 340 mg/m2 in pediatric patients fell within the 0.5 and 99.5 percentiles of the model-projected range scaled from adult measurements and the predicted plasma concentration-time profiles were generally in good agreement for the pediatric cohort (n = 67), except for subjects aged ≤2 years, for which the exposure appeared to be overpredicted. Nonetheless, the prediction was 1.5-fold of the adult value at 1 year of age. Of interest, the model was used to better understand the differences in prediction of children and adults, which seems to be the mixed effect of changing distribution volume and blood flow, in addition to clearance maturation with age. Overall, in conjunction with a population PK analysis, PBPK modeling suggested that the proposed posology of 340 mg/m2 in pediatric patients from 1 to 18 years of age was appropriate for imatinib.
Pediatric PBPK Modeling In-House at Epizyme
Building on many of the retrospective analyses described above and the recent regulatory precedent, we have applied PBPK modeling in a prospective manner to enable the first trials in pediatric patients for two first-in-class epigenetic therapies: pinometostat in mixed lineage leukemia–rearranged (MLL-r) and tazemetostat in integrase interactor (INI)-1–deficient solid tumors. The preemptive utilization of this approach early in development has informed some important aspects of phase I pediatric trial design, including starting dose regimen and blood sampling strategy. With model verification and optimization on availability of PK data in pediatric patients, the PBPK modeling workflow can be further applied to address additional clinical pharmacology questions such as drug interactions and PK/PD analysis, through the course of development of these investigational therapies in pediatric populations.
Relapsed/refractory MLL-r acute leukemia in children has a poor prognosis, with a reported 5-year overall survival for children aged <1 year with recurrent MLL-r leukemia being 4%–20%. The mixed lineage leukemia (MLL) gene, on chromosome 11q23, codes for a histone methyltransferase (HMT) that is responsible for methylation of H3K4, a modification associated with active transcription. Translocations of MLL result in the loss of the SET or catalytic domain of the protein with the most common translocation partners, AF4, AF9, and ENL, recruiting another HMT, the disruptor of telomeric silencing 1-like (DOT1L). The aberrant recruitment of DOT1L to MLL fusion target genes results in ectopic H3K79 methylation and increased expression of genes including HOXA9 and MEIS1, which are involved in leukemogenesis of MLL-r leukemias. Pinometostat (EPZ-5676) is a small molecule inhibitor of DOT1L with subnanomolar affinity and > 37,000-fold selectively against other HMTs. Preclinically, pinometostat selectively inhibits intracellular histone H3K79 methylation and downstream target gene expression and demonstrated complete tumor regressions in an MLL-r leukemia xenograft model (Daigle et al., 2013). Because of the unmet need of MLL-r in children, we employed a prospective PBPK modeling approach leveraging pinometostat preclinical data (Basavapathruni et al., 2014) and early clinical data from the first-in-human phase I open label study in adult patients with relapsed/refractory leukemia (Stein et al., 2014), to guide dose selection and trial design for a companion pediatric study (Waters et al., 2014; Shukla et al., 2015). A PBPK model describing the concentration-time profile of pinometostat after continuous intravenous administration in adult patients at dose levels of 24–90 mg/m2 per day was built using Simcyp (Simcyp Ltd., Sheffield, UK). The quantitative contribution of P450 enzymes to the metabolic clearance of pinometostat was based on intrinsic clearance (CLint) data derived from in vitro recombinant P450 systems using a relative activity factor approach. Pinometostat is a substrate for CYP3A4 and CYP2C19 in human and these enzymes are believed to account for 90% and 10% of the metabolic intrinsic clearance respectively. The metabolic intrinsic clearance was scaled using the well stirred model to generate a total hepatic in vivo CLint, which was then allocated to the P450 isoforms involved in the metabolism of pinometostat and thus allowed isoform-specific ontogeny functions to be used in the translation to the pediatric setting. The renal clearance in human was low (0.1 l/h) and was also incorporated into the model. Pinometostat was shown to preferentially bind to AAG in vitro (manuscript in preparation) and this allowed both the extent of binding and identity of plasma proteins involved to be included in the model, together with the maturation function for AAG. Early in model optimization, perfusion-limited kinetics were not able to recapitulate the plasma steady-state profile due to a marked overprediction of total clearance (several fold above the median observed clearance of 5.5 l/h). Using permeability-limited kinetics and a low steady-state volume of distribution (0.08 l/kg, derived from a separate compartmental analysis) was consistent with the short terminal half-life (t1/2) observed postinfusion and was able to fit the data well in terms of CL, steady-state plasma concentration, and the initial phase after the start of infusion. A sensitivity analysis of the passive diffusion clearance indicated that a value of 0.0075 mL/min/million hepatocytes gave the best model fit. This was plausible and consistent with the physicochemical properties and preclinical ADME data which showed low permeability in MDCK cell monolayers and a greater than 20-fold higher liver microsomal scaled CLint compared with hepatocyte scaled CLint (Basavapathruni et al., 2014). The adult model simulations and observed data are shown in Fig. 2A, with the Css predicted within 2-fold of the observed values and CL predicted within 20% of observed across the dose range 24–90 mg/m2 per day. Having built and qualified the PBPK model for pinometostat using adult PK data, the next step was to prospectively estimate exposures across the pediatric age range from 1 month to 18 years. The median clearance projected in pediatric patients ranged from 0.5 l/h in 1- to 3-month-olds to 4.2 l/h in 6- to 18-year-olds, and when normalized for body surface area (BSA) ranged from 1.8 l/h per square meter to 3.2 l/h per square meter, respectively. A modest 1.7-fold difference in BSA-normalized clearance between infant and adolescent suggested a dampening of the effect of P450 ontogeny on the clearance of pinometostat, as a direct result of age-independent, permeability-limited hepatic uptake incorporated into the model. This observation is in stark contrast to the clearance of prototypical CYP3A substrates in children which show an age-dependent relationship, concordant with the maturation of CYP3A expression during the first 2 years postnatal (Björkman, 2005; Hines, 2009). The model assumption with potential to affect predictive accuracy was clearly the permeability-limited metabolic clearance being independent of age. This was considered reasonable since the basis for the rate-limiting passive diffusion clearance of pinometostat was physicochemical in nature, as opposed to transporter-mediated active efflux, for example, which may show age dependent expression or activity. Simulations of the predicted steady-state systemic exposure of pinometostat in pediatric virtual populations were used to support starting dose selection. At a fixed BSA-normalized dose, adjustments of 55%–65% of the adult dose were projected in infants up to 2 years old to achieve equivalent exposures to adults; in children aged >6 years, the dose was predicted to be equivalent to the adult dose. For the practical purposes of trial conduct in this rare patient population, the starting dose level and age stratification was further simplified in a conservative manner to derive a pediatric starting dose of 80% and 50% of the highest adult dose (90 mg/m2 per day) in >12-month-olds and <12-month-olds, respectively. PK data recently obtained from this study (Shukla et al., 2015) have enabled model verification and this is summarized in Fig. 2, B and C. The observed Css at 70 mg/m2 per day (n = 6; age range, 1.25–15 years) was within 0.75- to 2.33-fold of the predicted Css across this age range, showing that the data were consistent with the postulated effect of dampened P450 ontogeny on the clearance of pinometostat. In this cohort, clearance ranged from 1.8 to 3.7 l/h per square meter, showing good concordance with the PBPK model CL projection of 2.4–3.2 l/h per square meter in subjects older than 1 year. The limited enrollment of patients less than 1 year of age in the relapsed/refractory setting precluded a more thorough assessment of P450 ontogeny on pinometostat clearance. Notwithstanding, the observed Css at 90 mg/m2/d (n = 5; age range, 1–15 years) showed a similar level of agreement with model predictions and was comparable with the plasma exposure observed in adult at this dose level (Fig. 2C; Shukla et al., 2015).

(A) Representative plot showing the model fit for interim adult PK data at 90 mg/m2 per day. Data at 24 mg/m2 per day (n = 5) and 36 mg/m2 per day (n = 3) showed similar fits. The black line represents the mean, the dotted line is the 95% confidence interval, and gray lines represent mean results from individual simulation trials (20 trials of four subjects). The majority of data fall within the 95% confidence interval of the model. (B) Representative plots of the simulated time-concentration profile for pinometostat administered at 70 mg/m2 per day in age groups ranging from 6 months to 18 years (solid gray lines), with observed plasma concentrations in pediatric patients receiving an infusion of 70 mg/m2 per day (open circles). Medians are shown as solid black symbols. (C) Box and whisker plot of the plasma Css of pinometostat administered at 45, 70, and 90 mg/m2 per day in pediatric patients (median age range, 0.33, 5.5, and 4 years, respectively). The dotted line corresponds to the observed median plasma Css in adults at 90 mg/m2 per day, and the shaded area represents the predicted Css range in pediatric patients aged >1 year at the dose level of 90 mg/m2 per day.
Tazemetostat (EPZ-6438) is a selective, orally active, small molecule inhibitor of the histone-lysine methyltransferase Enhancer of Zeste Homolog 2 (EZH2), which has been implicated in the pathogenesis of a variety of malignancies, including B-cell non-Hodgkin lymphoma and integrase interactor (INI)-1–deficient tumors (Knutson et al., 2013, 2014). INI-1–negative malignant rhabdoid tumors, for instance, occur mainly in young children. Other INI-1–negative tumors include epithelioid sarcomas, epithelioid malignant peripheral nerve sheath tumors, extraskeletal myxoid chondrosarcoma, renal medullary carcinoma, and myoepithelial carcinomas, characterized by their rarity and unmet medical need. INI-1–deficient tumors include several tumors for which there is no established standard of care, with a median survival of 0.3 years after recurrence in children. The primary objective of the PBPK modeling approach was to simulate tazemetostat exposures in adults prior to scaling to a pediatric population and to predict starting doses in children that would provide systemic exposures in the safe and efficacious range observed in adults. Tazemetostat PK data from patients enrolled in the dose escalation cohorts of the first-in-human phase I clinical study across the oral dose range of 100 mg (suspension) and 100, 200, 400, 800, and 1600 mg (tablet) twice daily, together with physicochemical and in vitro data including plasma protein binding, blood partitioning, metabolic stability, and P450 phenotyping, were used to simulate adult exposures by PBPK modeling using GastroPlus (version 8.5; Simulation Plus, Inc). In these adult patients, tazemetostat was rapidly absorbed with a maximum plasma concentration observed approximately 1 to 2 hours postdose. Plasma concentrations declined in a monoexponential manner with a mean t1/2 of approximately 3–6 hours. The tazemetostat AUC was slightly higher than dose proportional after a single dose, reasonably dose proportional at steady state, and comparable between the tablet and suspension formulations. After multiple dosing (day 15), the median time to reach maximum plasma concentrations (tmax) and t1/2 remained unchanged across the dose range, with a modest decrease in systemic exposure on day 15 and no further reduction onward (Ribrag et al., 2015). In the PBPK model, partition coefficients were set using the Lucakova (Rodgers–Rowland Single) equation for perfusion-limited tissues. Effective permeability was fitted to the clinical PK data in a top-down approach, refining the initial Cmax estimated from an in vitro permeability assay. Using permeability data from porcine kidney epithelial cells resulted in a 1.6-fold underestimation of Cmax at 800 mg. This difference likely reflects a concentration-dependent effect since the in vitro assay was performed at low micromolar concentrations, whereas the local gastrointestinal concentration could be as high as molar range (800 mg in 250 ml). Total clearance consisted of hepatic, intestinal, and renal components. Renal clearance was set as passive renal filtration (i.e., product of free fraction and glomerular filtration rate), which was in line with the preliminary estimate of renal clearance in the phase I study. Metabolic clearances in the gut and liver were based on system parameters for the expression levels of CYP3A4 in each gut compartment and the average expression of CYP3A4 in liver. Human liver microsome kinetics (Km and Vmax) were used as initial inputs to model tazemetostat hepatic clearance in humans, prior to refinement based on observed clinical data, and it was assumed that the fraction of the drug metabolized by CYP3A4 was close to unity. The refinement of Km and Vmax values was not substantial, being within 20% of the experimentally determined values in human liver microsomes. Although the tazemetostat accumulation ratio (AUCday15/AUCday1) was suggestive of a modest induction of metabolism, omitting to include CYP3A4 induction was not detrimental to the simulation of the steady-state (day 15) exposures (Fig. 3A). Because accumulation ratios remained modest throughout the dosing range (accumulation ratio of 0.8 to 0.5), and since the simulated concentration-time profile was very similar to the observed, CYP3A4 induction kinetics were not added to this model. This is a limitation of the model, which likely accounts for the nonoptimal capture of the time dependence of tazemetostat PK, particularly at higher doses (although predicted day 1 AUC and Cmax were within 2-fold of observed values at doses of ≤800 mg). Furthermore, the scaling of CYP3A autoinduction to a pediatric population (i.e., ontogeny of nuclear hormone receptors such as the pregnane X receptor) is not sufficiently understood to allow quantitative modeling. This would have made it difficult to account for autoinduction in the simulations of the pediatric population even if it had been included in the adult model. Similarly, because no transporter kinetic data were available at the time of this study and sufficient P-gp ontogeny data are lacking, the effect of potential efflux transport on tazemetostat PK was not considered in this model. Given these limitations, the potential effects of autoinduction should be considered in the interpretation of the model results. Based on visual inspection, the model fit of the adult exposures (n = 24) adequately described the time-concentration profiles of tazemetostat and resulted in prediction of mean AUCss and oral clearance (CL/F) within ±30% of the observed results across the dose range. In addition, mean Cmax,ss values were predicted within 2-fold of the observed values (Fig. 3B; Rioux et al., 2015). The resultant adult model was then used to simulate tazemetostat steady-state exposures in discrete pediatric age ranges (6 months to 1 year, >1–2 years, >2–6 years, >6–12 years, and >12–18 years) after twice-daily administration of an oral suspension. In addition to pediatric physiology, the simulations accounted for ontogeny in hematocrit, plasma protein levels, and P450 expression (liver and gut). Although CYP3A4 ontogeny was modeled, the propensity for CYP3A7-mediated metabolism of tazemetostat was unknown and so no interaction with this major fetal isoform was accounted for in the pediatric model. Nevertheless, the effect of potential metabolism by CYP3A7 is expected to be limited, since projection of pediatric doses was limited to patients aged ≥6 months (Hines, 2009). Using this exposure-based analysis, pediatric doses that afforded the target AUC (80% of adult AUCss at 800 mg or 390 mg/m2 twice daily) were identified. On a BSA-normalized basis, the projected doses were comparable across the age range (1–18 years), from 270 to 305 mg/m2 twice daily, with a slightly lower projected dose of 230 mg/m2 twice daily, for the group aged 6 months to 1 year. This lower dose was attributable to multiple factors, including CYP3A4 ontogeny (60%–80% of adult expression in the liver for the group aged 6 months to 1 year), change in free fraction, volume of distribution, and blood flow. By contrast, the tazemetostat fraction absorbed was not projected to change significantly from adults to children for doses of ≤300 mg/m2 twice daily. Because the projected doses by age were comparable, population simulations were performed to determine the corresponding exposures for each age range at two fixed doses (240 and 300 mg/m2 twice daily; Fig. 3B), since using an age-independent dose was expected to facilitate trial design in this rare patient population. In brief, at 300 mg/m2 twice daily, mean steady-state AUC0–τ values ranged from approximately 80% to 100% of that observed in adults at 800 mg twice daily. By contrast, the mean steady-state Cmax was projected to range from 110% to 190% of that observed at 800 mg twice daily in adults, but within the safe and efficacious exposure range defined in adults at doses up to 1600 mg twice daily. A lower dose of 240 mg/m2 twice daily was projected to maintain AUCss values ≤ 80% of the adult target exposure across the age range of 1–18 years.
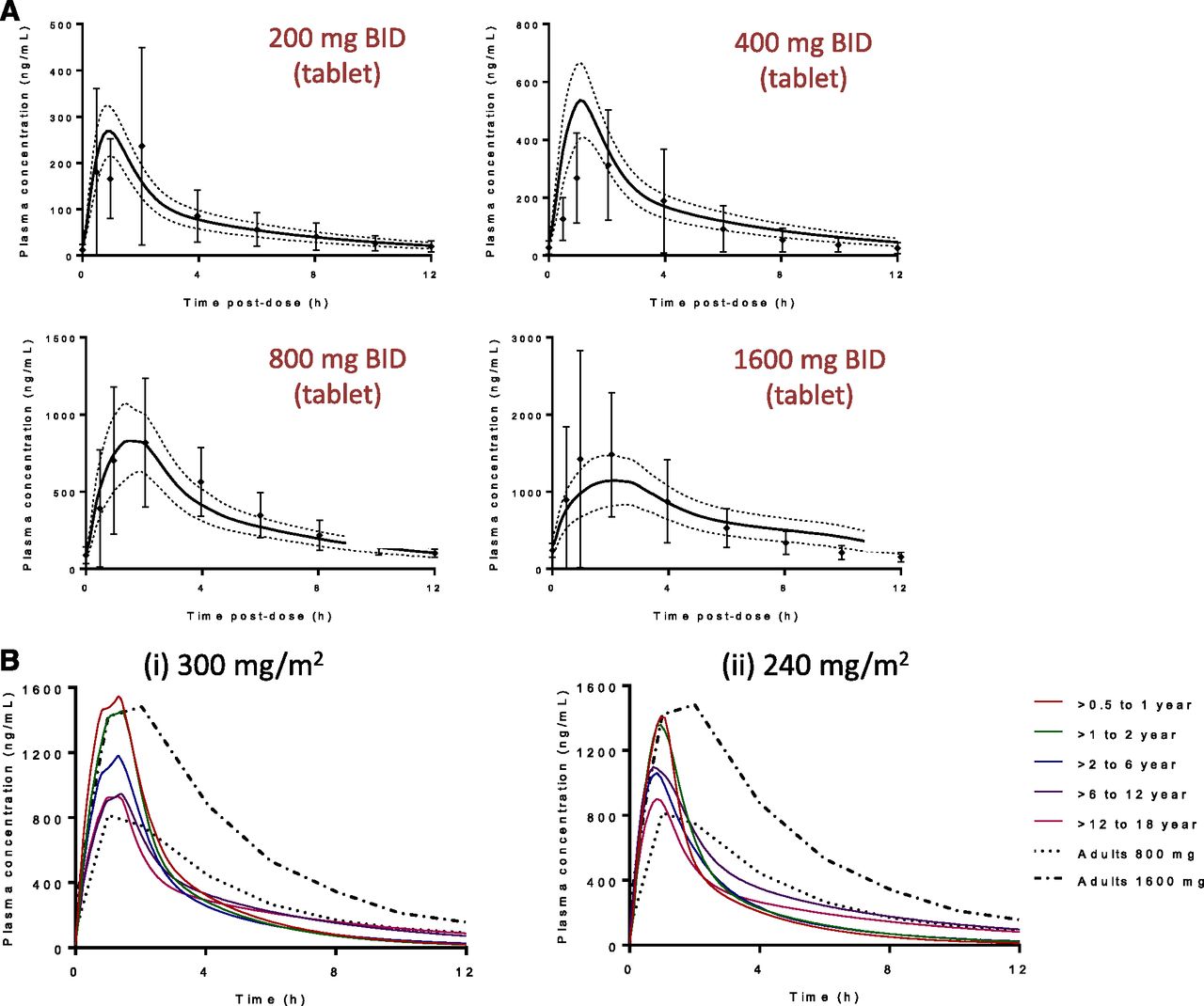
(A) Representative plots showing the model fit for tazemetostat interim adult PK data at steady state (day 15, n = 24) showed a good fit for each dose group. Symbols represent observed data from individuals (± S.D., n = 3 or 6 per dose) and the solid line represents the mean profiles predicted from the PBPK model in GastroPlus. Dotted gray lines represent the 90% confidence interval. (B) Predicted mean total steady-state plasma concentration-time profiles of tazemetostat administered as a 300-mg/m2 twice-daily or a 240-mg/m2 twice-daily oral dose across the age ranges, with mean measured total steady-state plasma concentration-time profile of tazemetostat administered as a 800-mg (390 mg/m2) or 1600-mg (780 mg/m2) twice-daily oral dose in adults (n = 6 per dose). BID, twice daily.
These studies demonstrate the prospective application of PBPK early in clinical development to support clinical trial design, and they have provided dosing recommendations for the ongoing trials of pinometostat and tazemetostat in pediatric patients.
Current Challenges and Limitations in Pediatric PBPK Modeling
Although multiple ontogenic physiologic processes are fairly well characterized in PBPK models, knowledge gaps remain because some growth and maturation trajectories are still only partially or poorly characterized. Among these, the Pediatric Biopharmaceutics Classification System Working Group determined that additional research was required to fully understand age-based changes in gastrointestinal fluid composition, motility, absorptive surface area, and pH ranges encountered along the gastrointestinal tract, all of which are critical factors needed to better understand age-dependent drug absorption (Abdel-Rahman et al., 2012).
For clearance, incomplete knowledge of the developmental patterns for some P450 isoforms, UGTs, and other conjugative enzymes remains a challenge. Partial coverage of the pediatric age spectrum, sparse data in extrahepatic tissues, and an undetermined fraction metabolized by CYP3A7 (a major isoform in neonates) are some of the factors that can make scaling adult clearance to children a complex endeavor (Kearns et al., 2003; Abdel-Rahman et al., 2012). In addition, the parameterization of P450 ontogeny functions used in PBPK models may be based on data derived either in vitro (expression and/or activity) or in vivo (activity only), and recent studies have highlighted the potential for marked differences between the two methods (Salem et al., 2014; Upreti and Wahlstrom, 2016). In parallel, understanding of the pharmacokinetic effect of the transporter-specific expression level in discrete age strata is still limited and remains a challenge when performing PBPK modeling for pediatric applications. Screening of the mRNA expression of uptake and efflux transporters in the human liver reveals that only a small number of isoforms are detectable up to 1 month after birth; in general, although expression increased with age, the timing at which adult mRNA levels were reached did vary (Klaassen and Aleksunes, 2010; Abdel-Rahman et al., 2012; Mooij et al., 2014). Although more data are becoming available on transporter expression levels, studies on transporter activity in pediatric populations are still scarce. As data highlighting the importance of drug transporters in adult medicine continue to emerge, this critical knowledge gap in the pediatric population becomes even more evident, as raised by the National Institutes of Health Pediatric Transporter Working Group in their recent white paper (Brouwer et al., 2015).
Although PBPK modeling is increasingly used for prediction of DDI potential in adult populations, there are a number of gaps in the robust prediction of DDI potential in pediatrics. From the perspective of a victim drug interaction, the magnitude of a change in metabolic clearance depends on the fraction metabolized by the inhibited or induced pathway(s), and age-related changes owing to ontogeny may affect the predictive accuracy in the quantitative contribution of individual clearance mechanisms (Salem et al., 2013). Furthermore, nuclear hormone receptor mRNA expression (e.g., the pregnane X receptor and the constitutive androstane receptor, known to regulate expression of genes involved in drug disposition) demonstrate considerable interindividual variability in human fetal and pediatric livers (Vyhlidal et al., 2006), with the effect on P450 induction not yet fully understood. This confounds modeling of complex drug interactions such as simultaneous inhibition and induction of metabolic pathways and/or transporters by a drug (and/or its metabolites), presenting significant challenges to the prediction of clinical DDIs in adults (Varma et al., 2015) and even more so in pediatric populations.
Ultimately, the ability to model the effect of disease state on PK would further enhance the PBPK modeling approach by factoring in the physiologic sequelae of cancer in children relative to the current databases for healthy pediatric subjects; this is not currently a surmountable task, given the pleiotropic nature of malignant neoplastic disease. Furthermore, the extension of pediatric PBPK modeling to include a pharmacodynamics component remains an area of much-needed research.
The evaluation of the predictive performance of published PBPK models is an area that is in need of further scrutiny in moving toward a more standardized and systematic reporting of PBPK modeling and simulation efforts, and this issue has been raised by a number of groups from academia, industry, and regulatory authorities (Maharaj and Edginton, 2014; Zhao, 2014; Sager et al., 2015).
Summary and Conclusions
PBPK modeling and simulation in pediatric oncology is still quite nascent, limited to a few well characterized drugs and often performed as part of a retrospective analysis validating the methodology in this area of drug research. Nevertheless, these studies demonstrated the ability of PBPK modeling to simulate PK in pediatric patients, refine clinical trial design, inform on covariates influencing drug exposure levels in children, and evaluate the potential for clinically relevant DDIs. The case studies presented for pinometostat and tazemetostat provide further validation of their prospective utility a priori to the first-in-pediatrics clinical investigation, providing guidance on starting dose regimen and judicious selection of blood sampling times. Modeling and simulation endeavors in this area of drug research are particularly challenging since the patient population is rare. First-in-pediatric study populations tend to be relatively small, which impedes the statistical rigor usually associated with evaluating age as a covariate in later-stage trials. However, as detailed herein, PBPK modeling applied with a fit-for-purpose tenet can enable informed decisions on the starting dose in phase I pediatric oncology trials, in an effort to ensure that the age continuum is treated with a starting dose at or close to a safe and efficacious one. The future direction of PBPK modeling in the field of pediatric oncology drug development is likely to see expanded application in early clinical development with the assessment of DDI potential, influence of disease covariates and providing mechanistic insight into observed variability in the target patient population.
Acknowledgments
The authors thank Epizyme colleagues, clinical investigators, and their teams and, most importantly, the patients and families who participated in the studies reported herein.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Rioux, Waters
Footnotes
Abbreviations
- AAG
- α-1-acid glycoprotein
- ADME
- absorption, distribution, metabolism, and excretion
- AUC
- area under the curve
- AUCss
- area under the curve at steady state
- BSA
- body surface area
- CL/F
- oral clearance
- Cmax
- peak serum concentration
- CLint
- intrinsic clearance
- DDI
- drug–drug interaction
- DME
- drug-metabolizing enzyme
- DOT1L
- disruptor of telomeric silencing 1-like
- EPZ-5676
- 9-[5-deoxy-5-[[cis-3-[2-[6-(1,1-dimethylethyl)-1H-benzimidazol-2-yl]ethyl]cyclobutyl](1-methylethyl)amino]-β-d-ribofuranosyl]-9H-purin-6-amine
- EPZ-6438
- N-[(1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-5-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-4-methyl-4′-(4-morpholinylmethyl)-[1,1′-biphenyl]-3-carboxamide
- FDA
- Food and Drug Administration
- GST
- glutathione-S-transferase
- HMT
- histone methyltransferase
- INI
- integrase interactor
- MLL
- mixed lineage leukemia
- MLL-r
- mixed lineage leukemia–rearranged
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetics
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- t1/2
- terminal half-life
- UGT
- uridine diphosphoglucuronosyl transferase
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}