


Abstract
Multidrug resistance (MDR) is a common limitation for the clinical use of microtubule-targeting chemotherapeutic agents, and it is the main factor for poor prognoses in cancer therapy. Here, we report on deoxypodophyllotoxin (DPT), a promising microtubule inhibitor in phase 1, as a promising candidate to circumvent this obstacle. DPT remarkably suppressed tumor growth in xenograft mice bearing either paclitaxel (PTX)-sensitive MCF-7/S or acquired resistance MCF-7/Adr (MCF-7/A) cells. Also, DPT exhibited similar accumulation in both tumors, whereas PTX displayed much a lower accumulation in the resistant tumors. In vitro, DPT exhibited a much lower resistance index (0.552) than those of PTX (754.5) or etoposide (38.94) in both MCF-7/S and MCF-7/A cells. Flow cytometry analysis revealed that DPT (5 and 10 nM) caused arrest of the G2/M phase in the two cell lines, whereas PTX (up to 10 nM) had no effect on cell-cycle progression of the MCF-7/A cells. Microtubule dynamics assays revealed that DPT destabilized microtubule assembly in a different mode. Cellular pharmacokinetic assays indicated comparable intracellular and subcellular accumulations of DPT in the two cell lines but a much lower retention of PTX in the MCF-7/A cells. Additionally, transport assays revealed that DPT was not the substrate of P-glycoprotein, breast cancer resistance protein, or MDR-associated protein 2, indicating a lower occurrence rate of MDR. DPT might be a promising microtubule inhibitor for breast cancer therapy, especially for treatment of drug-resistant tumors.
Introduction
Microtubules are composed mainly of α- and β-tubulin heterodimers, which can be bound by two groups: microtubule-stabilizing agents and microtubule-destabilizing agents. As reported, microtubule-binding agents are involved in numerous fundamental intracellular processes, such as the most recognized, mitosis (Howard and Hyman, 2003); thus, they receive a great deal of attention in the treatment of cancer. Antimicrotubule drugs are one type of the most effective drugs in the treatment of breast cancer, ovarian cancer, and other tumors; however, the occurrence of multidrug resistance (MDR) is a common limitation to their clinical use. Paclitaxel (PTX), a microtubule-stabilizing agent approved by the Food and Drug Administration for breast and lung cancer treatment, has demonstrated to be the P-glycoprotein (P-gp) efflux pump substrate that causes increased efflux (Gottesman, 1993) and consequently reduces paclitaxel’s intracellular concentration and develops drug resistance to the breast cancer therapy (Dumontet and Jordan, 2010). Therefore, novel microtubule inhibitors with high efficacy to MDR tumors are greatly needed.
Deoxypodophyllotoxin (DPT), isolated from Anthriscus sylvestris, is an active component that has potent antiproliferative and antitumor effects against a broad variety of cancer types by modulating the microtubule (Shin et al., 2010; Khaled et al., 2013, 2016; Kim et al., 2013; Wang et al., 2015b). As a promising microtubule inhibitor, DPT and its intravenous formulation of β-cyclodextrin inclusion complex (Zhu et al., 2010) have been adopted for phase 1 evaluation. A physiologically based pharmacokinetics model was also established for better efficacy and safety assessment (Chen et al., 2016). Previous studies have demonstrated that DPT exerts an antitumor effect on the human BC MDA-MB-231 cell line in vivo and in vitro (Benzina et al., 2015; Khaled et al., 2016); however, the effect of DPT on drug-resistant cancer cells has yet to be investigated. Whether DPT treatment can induce drug efflux transporters and subsequently lead to MDR warrants further confirmation.
From macrocosm to microcosm, intracellular pharmacokinetics has attracted great attention to the monitoring of intracellular target binding and the intracellular disposition process of drugs (Tulkens, 1990; Krishan et al., 1997), including drug uptake, distribution, metabolism, and efflux within the cellular environment. In recent studies, doxorubicin (Adriamycin) resistance in MCF-7/A cells was identified by exploring the cellular pharmacokinetic mechanism, which could be reversed by inhibiting P-gp (Zhang et al., 2012). Therefore, intracellular pharmacokinetics has provided a new perspective (Zhou et al., 2011) to determine the underlying mechanisms of MDR, thus promoting efficacy in drug discovery to overcome MDR.
In the current study, we investigated the antitumor effect of DPT on both drug-sensitive and drug-resistant MCF-7 cell lines in vivo and in vitro. We also explored the discrepancy between PTX and DPT on MDR from the perspective of intracellular pharmacokinetics. In parallel, the potential of DPT to overcome MDR was further elucidated.
Materials and Methods
Reagents.
DPT (purity >99%) and DPT-HP-β-CD (content 2.8%) were provided by the Medicinal and Chemical Institute, China Pharmaceutical University (Chen et al., 2016). Paclitaxel (injection) was purchased from the Yangtze River Pharmaceutical Group (Jiangsu, China). Etoposide, digoxin, PTX, and verapamil were bought from Sigma-Aldrich (St. Louis, MO). Lovastatin was obtained from the Chinese National Institute for the Control of Pharmaceutical and Biologic Products (Beijing, China). Acetonitrile and methanol (Merck, Darmstadt, Germany) were of high-performance grade. Acetic acid and ammonium acetate of high-performance liquid chromatography grade were purchased from Adamas Reagent Co., Ltd (Shanghai, China). Analytical-grade sodium acetate was from Nanjing Chemical Reagent Co., Ltd (Jiangsu, China).
Animals.
Female athymic BALB/c nude mice (9 weeks old, 18–22 g) were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China). The mice received 5 × 106 exponentially growing MCF-7/S and MCF-7/A cells subcutaneously in the right flank, and estrogen pellets were implanted before injection. Before the experiment, the mice were housed 10 per cage in constant temperature and humidity with an automatic day/night rhythm (12-hour cycle) in a specific pathogen free-grade environment. When the tumors reached 100 mm3, the mice were randomly divided into three groups, followed by antitumor treatment. Before the experiment, the mice fasted overnight (12 hours) with free access to water. The Guidelines for Care and Use of Laboratory Animals and the protocols approved by the corresponding Animal Ethics Committee of China Pharmaceutical University (Nanjing, China) were followed throughout all the animal experiments.
Antitumor Activity of DPT and PTX in Tumor Xenograft Models.
Mice bearing MCF-7/S and MCF-7/A subcutaneous tumors in their right flank regions were randomly assigned to three groups (10 and 9 mice per group, respectively): 1) control group (saline, intravenous), 2) DPT (12.5 mg/kg, i.v.), and 3) PTX (12.5 mg/kg, i.v.). Mice in each group were injected once every 3 days and weighed, and the major (1) and minor (2) axes of tumors were monitored daily. Tumor volume was calculated according to the following formula: tumor volume (cubic millimeter) = 1/2 × a × b2. The survival ratio of the two models was >75%, so at least six mice in each group were guaranteed to be alive 10 days later. After 10 days, the mice were killed by CO2 asphyxiation, and the plasma, xenografts, heart, liver, spleen, lung, and kidney were collected. The rate of growth was calculated using eq. 1:
 (1)
(1)Cell Culture.
Human breast cancer cells MCF-7 (MCF-7/S), purchased from the American Type Culture Collection (Rockville, MD), and their acquired resistant cells (MCF-7/A, by long-term doxorubicin induction) were provided by the Institute of Hematology and Blood Diseases Hospital (Tianjin, China). MCF-7/A cells have been verified to overexpress P-gp compared with MCF-7/S cells, and they exhibit MDR (Zhang et al., 2012). These two cell lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and 100 U ⋅ ml−1 of penicillin and streptomycin (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2. CaCO2 cells, parent Madin-Darby canine kidney epithelial cells (MDCK), and MDR1-transfected MDCK cells (MDR1-MDCK) were obtained from ZheJiang University (Hangzhou, China) (Cao et al., 2011). Cells were cultured in Dulbecco’s modified Eagle’s medium, along with 10% fetal bovine serum and 100 U·ml−1 penicillin and streptomycin.
Cytotoxicity Assay.
The efficacy of PTX and DPT on MCF-7/S and Adriamycin-resistant MCF-7/A cells was determined by cell growth inhibition via 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide colorimetric assay after incubation with PTX and DPT under different concentrations. The concentration required to inhibit growth by 50% (IC50 values) were calculated from the survival curves using the Bliss method (Bliss, 1939; Zhang et al., 2012; Lu et al., 2015).
Cell-Cycle Analysis.
Cell-cycle distribution was addressed by determining the DNA content of the cells. Cells were fixed at 4°C in ethanol overnight and then resuspended in staining solutions containing RNase A (100 µg/ml) and propidium iodide (20 µg/ml) for 30 minutes. Finally, the DNA content was monitored by flow cytometry (FACS Calibur; BD, Franklin Lakes, NJ) and analyzed with CELLQUEST software (Becton-Dickinson, San Jose, CA).
Cellular Retention Assay.
The cells from passages 10 to 20 were seeded on 24-well cell culture plates. When 90% confluency was reached, the cells were treated with DPT or PTX (5 µM) for 15, 30, 60, and 120 minutes. After 2 hours of incubation, the cells were lysed via three freeze-thaw cycles. As for the efflux assay, the cells were incubated with DPT or PTX (5 µM) for 2 hours, and then the intracellular retention of the two drugs with or without verapamil (20 µM) was determined after 0, 15, 30, 60, and 120 minutes. Meanwhile, the protein concentrations were measured by the Bradford method. The concentration of DPT was determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS), and PTX was monitored as previously described (Liu et al., 2017). All the experiments were conducted in triplicate. The area under the curve of the intracellular accumulation was expressed with the average concentration of DPT or PTX after 2 hours of incubation.
Subcellular Distribution of DPT and PTX.
MCF-7 cells from passages 10 to 20 were cultured in 75 cm2 cell culture flasks. Upon reaching 90% confluency (>107 cells/flask), the cells were treated with PTX or DPT (1 µM). After 15, 30, 45 minutes, 1 hour, and 2 hours of incubation, the cytoplasm, mitochondria, and nuclei were extracted according to the instructions of the KeyGen Mitochondria/Nuclei isolation kit (Nanjing KeyGen Biotech. Co., Ltd., Nanjing, China) as described previously (Zhang et al., 2012). Meanwhile, the protein content of the cytoplasm, mitochondria, and nuclei was determined as described. Then, the concentration of DPT and PTX in each subcellular compartment was measured by LC-MS/MS, and it was further adjusted in terms of the original dosing volume. The area under the curve of the subcellular accumulation was expressed with the average concentration of DPT or PTX after incubation for 2 hours.
Tubulin Polymerization Assay.
The experiment was performed according to a tubulin polymerization assay kit (Cytoskeleton, Denver, CO). Briefly, the standard polymerization reaction containing 3 mg/ml tubulin in G-PEM (80 mM PIPES, 0.5 mM EGTA, 2 mM MgCl2, 1 mM GTP, and 10% glycerol, PH 6.9) was mixed with different compounds in a 96-well plate. Polymerization was started by incubation at 37°C and followed by absorbance readings at 340 nm every minute for 1 hour. All the experiments were repeated three times.
Transport Assays.
Parent and MDR1-transfected MDCK epithelial cells from passages 10 to 20 were seeded at 5 × 105 cells/1.12 cm2 density on Millipore Millicell inserts (12-mm diameter, 0.4-µm pore size; Millipore Corporation, Bedford, MA) in 24-well tissue culture plates. Transport studies were conducted 5 days after seeding. Briefly, the MDCK and MDR1-MDCK cells were gently rinsed with Hanks’ balanced salt solution followed by incubation for 20 minutes at 37°C. To evaluate the effect of P-gp inhibitor on the transport of DPT, verapamil (20 µM) was added to either the apical or basolateral side of the monolayer 30 minutes before the addition of DPT (0.5 and 1 µM) or digoxin (5 µM) for 2 hours. The apparent permeability values (Papp) were calculated according to eq. 2 (Ranaldi et al., 1996; Zhang et al., 2010; Poirier et al., 2014; Kodama et al., 2016): (2)where dQ/dt represents the slope of the transporter cumulative amount within the time course studied, A is the surface area of the Millicell inserts, and C0 is the initial concentration. The efflux ratio (ER) was obtained by calculating the ratio of Papp B-A to Papp A-B. A compound with a ratio >2.0 was qualified as a substrate of the efflux mechanism.
(2)where dQ/dt represents the slope of the transporter cumulative amount within the time course studied, A is the surface area of the Millicell inserts, and C0 is the initial concentration. The efflux ratio (ER) was obtained by calculating the ratio of Papp B-A to Papp A-B. A compound with a ratio >2.0 was qualified as a substrate of the efflux mechanism.
Histopathological Examination.
Tumor tissues were collected and processed for histopathological examination. Briefly, tumors were fixed in 4% paraformaldehyde and then embedded in paraffin wax. Sections were subsequently prepared at a thickness of 4 µm and stained with H&E. The percentage of the lesion area was calculated according to five random images from the sections of the individual mice captured by light microscope (Zeiss, Oberkochen, Germany).
LC-MS/MS Method for DPT Quantification.
Briefly, a 50-µl aliquot of the cell sample was precipitated with 150 µl of acetonitrile (containing lovastatin as the internal standard). After centrifugation, 10 µl of the supernatant was injected into the SCIEX 5500 system (Framingham, MA) with a Waters C18 column (3.0 × 50 mm, 2.5 µm; Waters, Milford, MA). The column and autosampler tray temperatures were designated as 40 and 4°C, respectively. Mobile phase A used 0.1% (v/v) acetic acid and 2 mM ammonium acetate in water, and mobile phase B used acetonitrile; A gradient 0 minute; 30% B → 2 minutes, 70% B → 4 minutes, 70% B → 6 minutes, 30% B → 7 minutes, 30% B → 8 minutes. The flow rate was 0.2 ml/min. The mass spectrometer was operated in the positive electrospray ionization mode. MS parameters were set as: ion source gas 1 as 55, ion source gas 2 as 35, curtain gas as 15, collision gas as medium, temperature 550°C, and ion spray voltage: 5.5 kV in the positive mode. Quantification was performed using the selected reaction monitoring mode: m/z 399.2→157 with declustering potential 85 V, and collision energy 35 V for DPT, m/z 405 → 199 with DP 75 V and CE 36 V for lovastatin. To ensure reliability, we evaluated the method via linearity, recovery, and matric effect, precision, and accuracy, and stability validation, which were in line with the bioanalytical recommendations (Supplemental Tables). The concentration of DPT in mice plasma and tissues were monitored as previously described (Yang et al., 2014) with quality control involved.
Statistical Analysis.
All the data are presented as mean ± S.D. Significance was determined between two groups using the Student’s t test. Multiple comparisons were analyzed with one-way analysis of variance. Representative graphs were made using GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA), and P values <0.05 were considered statistically significant.
Results
DPT Inhibited the Tumor Growth of Both MCF-7/S and MCF-7/A Xenograft Mice In Vivo.
We first established two xenograft models of drug-sensitive and drug-resistant human breast cancer (MCF-7/S and MCF-7/A, respectively) to investigate the toxic effect of DPT in vivo. DPT was intravenously (12.5 mg/kg) administered every 3 days. Concomitantly, the comparison was conducted with the same dose of PTX every 3 days. Ten days after the last administration, as shown in Fig. 1, A and E, both DPT and PTX exerted remarkable suppression of MCF-7/S xenograft growth. The average tumor weight of the MCF-7/S xenografts treated with DPT and PTX was 0.26 and 0.28 g, respectively, compared with that of the vehicle-treated animals of 0.56 g (Fig. 1C). Figure 1G revealed the inhibition of tumor volume growth of the MCF-7/S xenografts when supplemented with DPT (49.62%); PTX caused the inhibition of tumor volume growth of 53.86%.
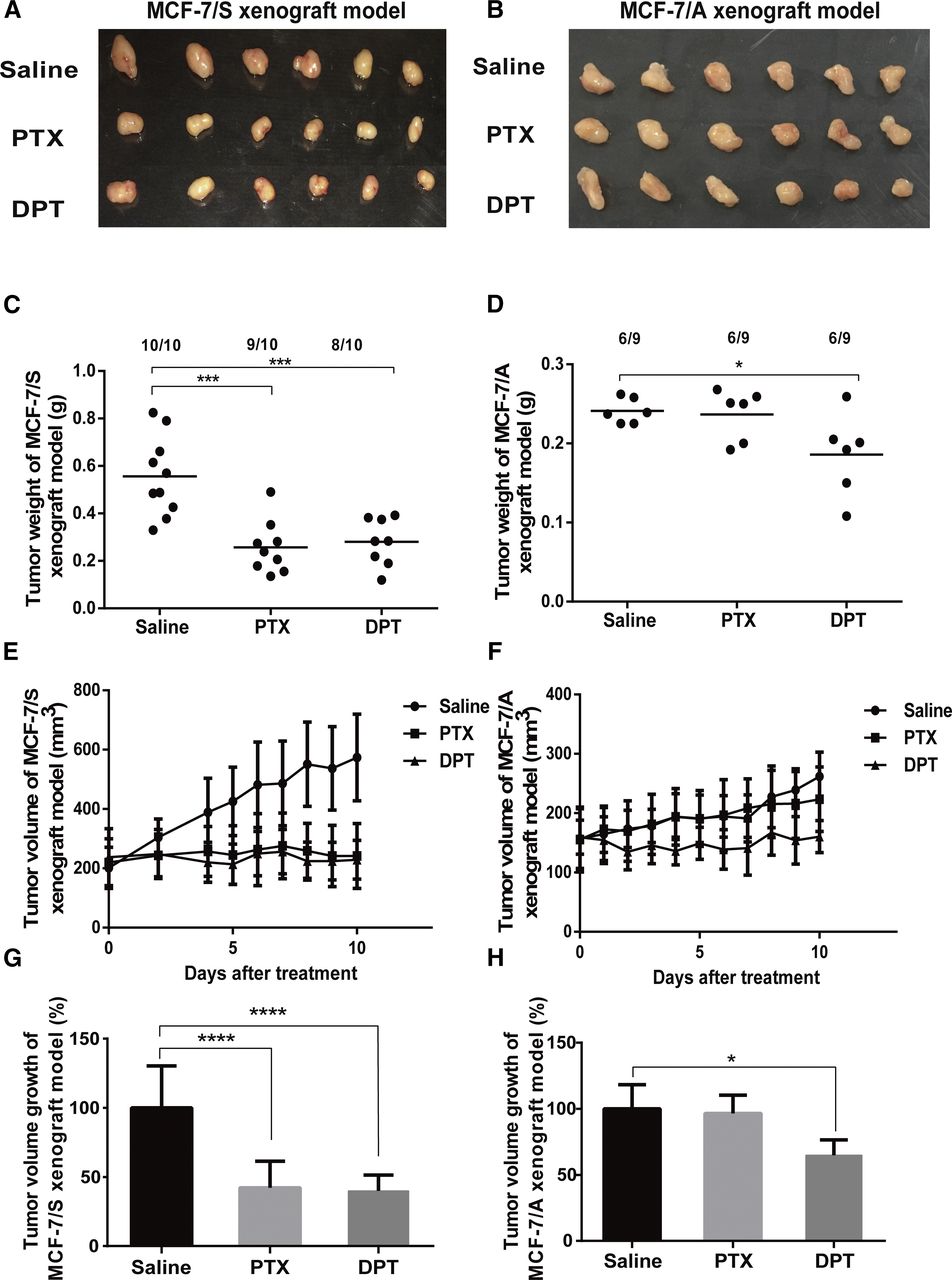
DPT inhibited the growth of MCF-7/S and MCF-7/A tumors in nude mice. Representative photos of the xenografts excised from MCF-7/S (A) and MCF-7/A (B) xenograft models are presented. The average weight of the xenografts excised from MCF-7/S (C) and MCF-7/A (D) xenograft models is shown. The average volume of the MCF-7/S (E) and MCF-7/A (F) xenografts in nude mice were both inhibited by DPT, with dosing schedules following every 3 days. In contrast, PTX exerted no effects on the growth of the MCF-7/A xenografts, with dosing schedules following every 3 days. A Vernier caliper was used to measure the tumor diameter serially, followed by the calculation of the relative tumor volume using the equations expressed in the Materials and Methods section. The percentage of tumor volume growth of the MCF-7/S (G) and MCF-7/A (H) xenograft models was expressed under the treatment of DPT and PTX. Data are expressed as mean ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. control.
As displayed in Fig. 1, B and F, DPT also showed a palpable reduction of the tumor growth of the MCF-7/A model, with the tumor weight of 0.17 g in contrast with that of the control group of 0.24 g (Fig. 1D). It was noteworthy that PTX showed just a marginal inhibitory rate (Fig. 1H), with the tumor weight of 0.24 g.
Additionally, no obvious body weight loss or abnormal behavior was observed in any of the DPT-treated groups; however, we found a notable loss of body weight in the PTX-treated MCF-7/A xenograft models. All the results demonstrated that DPT was effective in the treatment of both sensitive and drug-resistant human breast cancer and showed no obvious toxicity. Conversely, PTX exerted a low tumor inhibition rate, suggesting that PTX might develop resistance to the MCF-7/A cell lines.
DPT Induced Tumor Necrosis in Both the MCF-7/S and MCF-7/A Xenografts.
Histopathological examination of the xenografts was also performed in the MCF-7/S and MCF-7/A xenograft models. As shown in Fig. 2, the tumor cells grew vigorously and arranged irregularly with more chromatin in the control group; however, under the treatment of DPT, the lesion areas of the MCF-7/S and MCF-7/A xenografts were 68% ± 7% and 25% ± 12%, respectively, within which, the sparsely arranged tumor cells were diffusely distributed with a pale cytoplasm, polymorphous nuclei, and necrosis; mitotic activity was absent. In sharp contrast to DPT, PTX exerted no effects on the MCF-7/A xenografts.
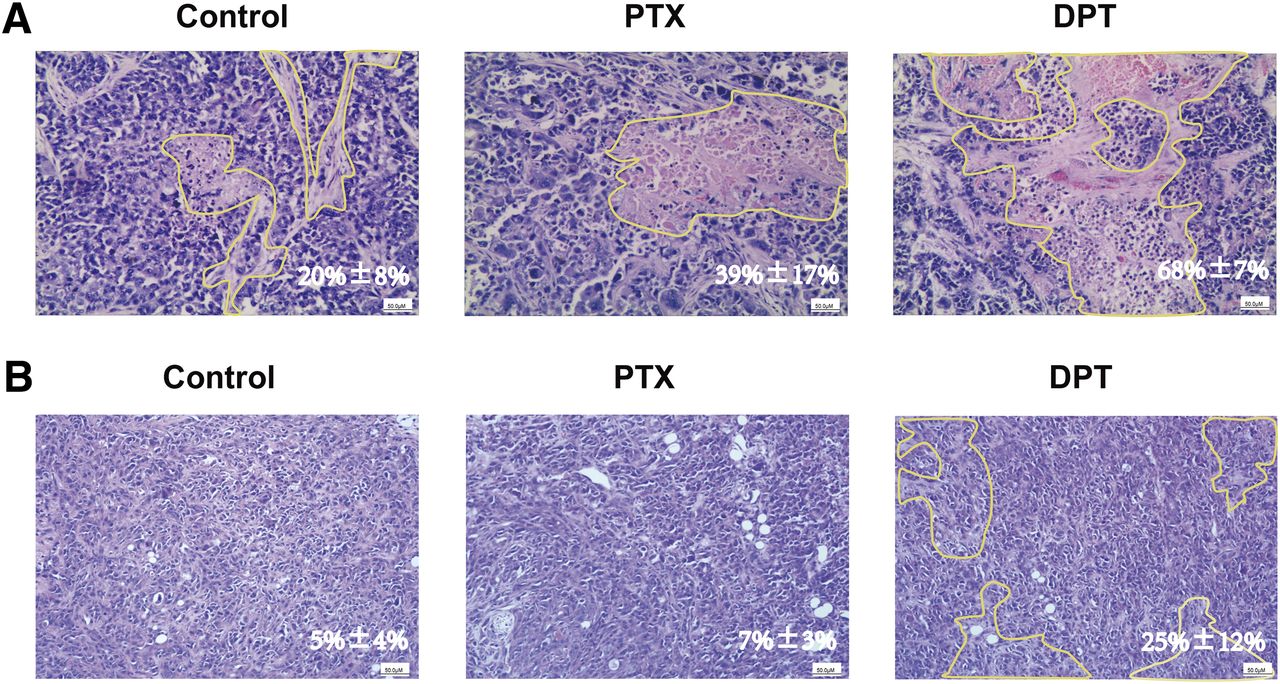
DPT supplementation in the MCF-7/S (A) and MCF-7/A (B) xenograft models increased nuclear pleomorphsim and necrosis. Tumors excised from the xenograft models were fixed with formalin and sectioned for H&E staining with the original magnification, 100×.
DPT Displayed Comparable Biodistribution in the MCF-7/S and MCF-7/A Xenograft Models.
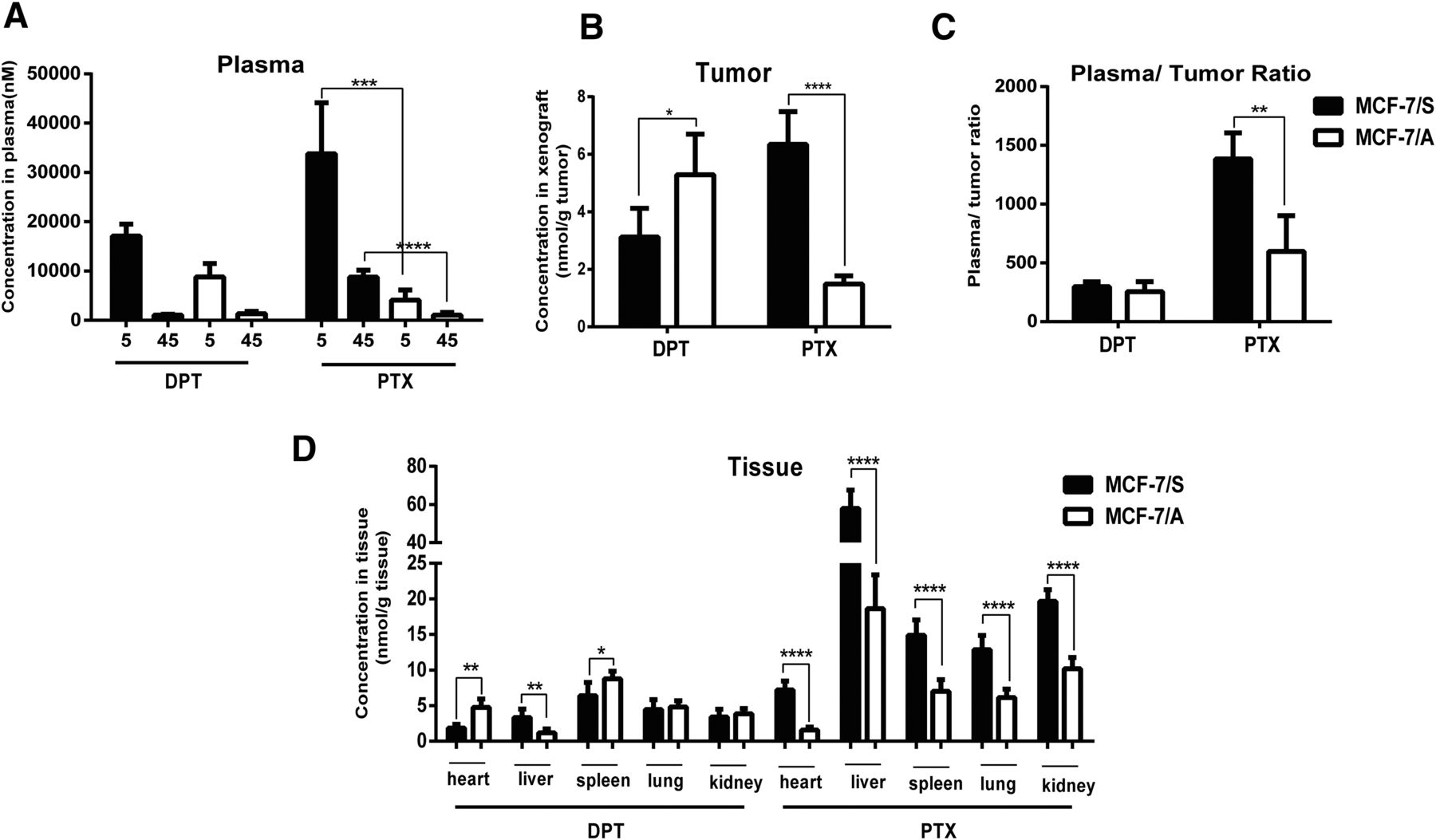
In support of the notion that DPT was more effective in the treatment of MCF-7/A xenografts, we next monitored the biodistribution of DPT and PTX in both the MCF-7/S and MCF-7/A xenograft models. The drug concentrations in the plasma were determined at 5 and 45 minutes after DPT and PTX administration (Fig. 3A). Subsequently, the concentrations of DPT and PTX in the xenografts were monitored at 45 minutes. As presented in Fig. 3B, the concentration of DPT in the MCF-7/A xenografts was a little higher than that in the MCF-7/S xenografts, along with a parallel plasma/tumor ratio (Fig. 3C). Conversely, in contrast to the MCF-7/S xenografts, the concentration of PTX was decreased by 4.26-fold in the MCF-7/A xenografts, and the plasma/tumor ratio was decreased by 2.15-fold. Meanwhile, PTX was distributed less in other nonmalignant tissues (Fig. 3D).

Biodistribution of DPT and PTX in the MCF-7/S and MCF-7/A xenograft models. (A) Concentration-time curve of DPT and PTX in the plasma of the MCF-7/S and MCF-7/A xenograft mice. (B) The concentration of DPT and PTX in tumors excised from MCF-7/S and MCF-7/A xenograft mice. (C) The plasma/tumor ratio of DPT and PTX in the MCF-7/S and MCF-7/A xenograft mice, calculated by the average concentration of DPT or PTX in the plasma versus that in the tumor at the designated time (45 minutes after injection). (D) Drug biodistribution of DPT and PTX in nonmalignant tissues from the heart, liver, spleen, lung, and kidney of the MCF-7/S and MCF-7/A xenograft mice. Data are presented as mean ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus control.
DPT Exhibited a Much Lower Resistance Index than that of PTX or Etoposide in the MCF-7/S and MCF-7/A Cells.
Based on these findings, we next investigated the toxic effect of DPT on breast cancer cells. We chose MCF-7/S and their derivative MCF-7/A cells treated with different drugs at various concentrations to measure the cellular viabilities. As shown in Table 1, the IC50 values of DPT on the MCF-7/S and MCF-7/A cells were 10.61 and 5.86 nM, respectively, which showed that the MCF-7/A cells were sensitive to the effects of DPT. In contrast, with resistance index of 754.5 and 38.94, respectively, PTX and etoposide exerted decreased efficacy on the MCF-7/A cells.
IC50a values of DPT, PTX, and etoposide in MCF-7/S and MCF-7/A cells
DPT Induced G2/M Cell-Cycle Arrest in Both MCF-7/S and MCF-7/A Cells.
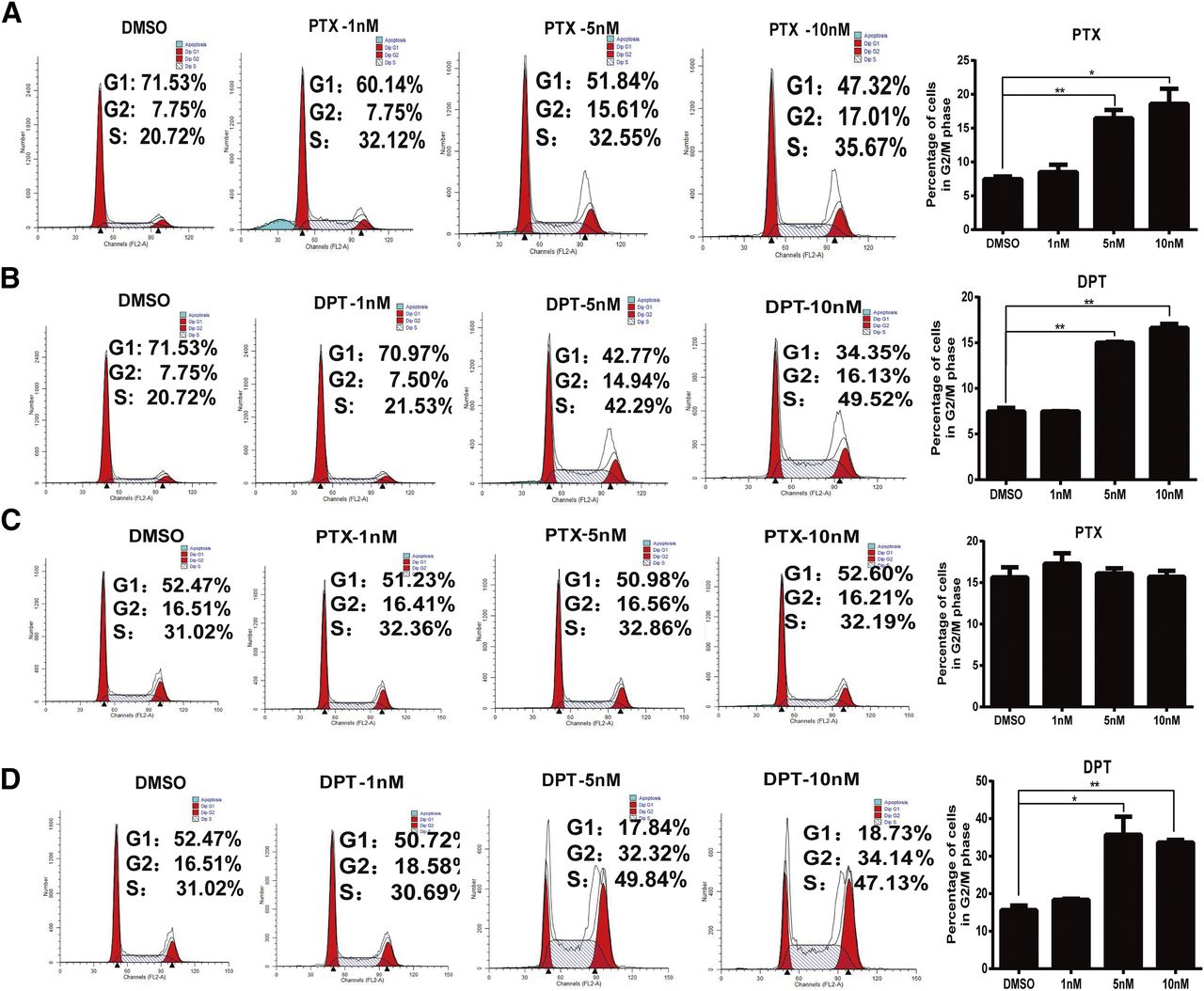
Generally, cell-cycle progression is strongly associated with tubulin polymerization. Inhibition of tubulin polymerization has been implicated in G2/M cell-cycle arrest in numerous cancer cell lines. We then compared the effect of DPT and PTX on cell-cycle progression of the MCF-7/S and MCF-7/A cells by flow cytometry. As depicted in Fig. 4, B and D, treatment with DPT (5 and 10 nM) caused a remarkable accumulation in the G2/M phase in dose-dependent manners with a concomitant decrease in the G1 phase in both the MCF-7/S and MCF-7/A cells. DPT induced obvious G2/M arrest at 5 nM. The percentage of MCF-7/S and MCF-7/A cells in the G2/M phase was 14.94% and 32.32%, respectively. In stark contrast to DPT, although PTX exerted a similar efficacy on the MCF-7/S cells, no induction was observed on the MCF-7/A cells (Fig. 4, A and C). These results indicated that DPT might be more active than PTX in the treatment of drug-refractory tumors, especially those with resistance to PTX.

Compared with PTX, DPT induced G2/M cycle arrest in the MCF-7/S and MCF-7/A cells. (A and B) PTX and DPT caused G2/M phase arrest in the MCF-7/S cell at concentrations of 1, 5, and 10 nM for 12 hours. (C) PTX did not induce cell cycle progression in the MCF-7/A cells. (D) DPT arrested the MCF-7/A cells at the G2/M phase at the specified concentrations of 1, 5, and 10 nM. Histograms represent the percentage of cell-cycle distribution at the G2/M phase. Data are presented as mean ± S.D. of three independent experiments. *P < 0.05; **P < 0.01 versus control (0 nM).
DPT Destabilized Microtubule Assembly in a Different Mode from that of PTX.
It has been reported that PTX can bind to the β-tubulin and stabilize microtubules. To determine the efficacy of DPT and PTX, we conducted the turbidity assay by recording the absorbance of the purified tubulin at 340 nM after incubation with PTX and DPT. The results indicated the antitubulin polymerization activities of DPT in a concentration-dependent manner as shown by the absence of the polymerized tubulin. In contrast, PTX resulted in a distinctive stabilization of microtubules as expected (Fig. 5).

Effect of DPT and PTX on tubulin polymerization. The efficacy of DPT (1, 2, 5, 10, and 20 µM) and PTX (10 µM) on tubulin polymerization was evaluated by turbidity changes recorded at a wavelength of 340 nm. The experiment was repeated three times. Data from the representative experiment are shown.
Cellular and Subcellular Accumulation of DPT in MCF-7/A Cells Is Similar to that in MCF-7/S Cells.
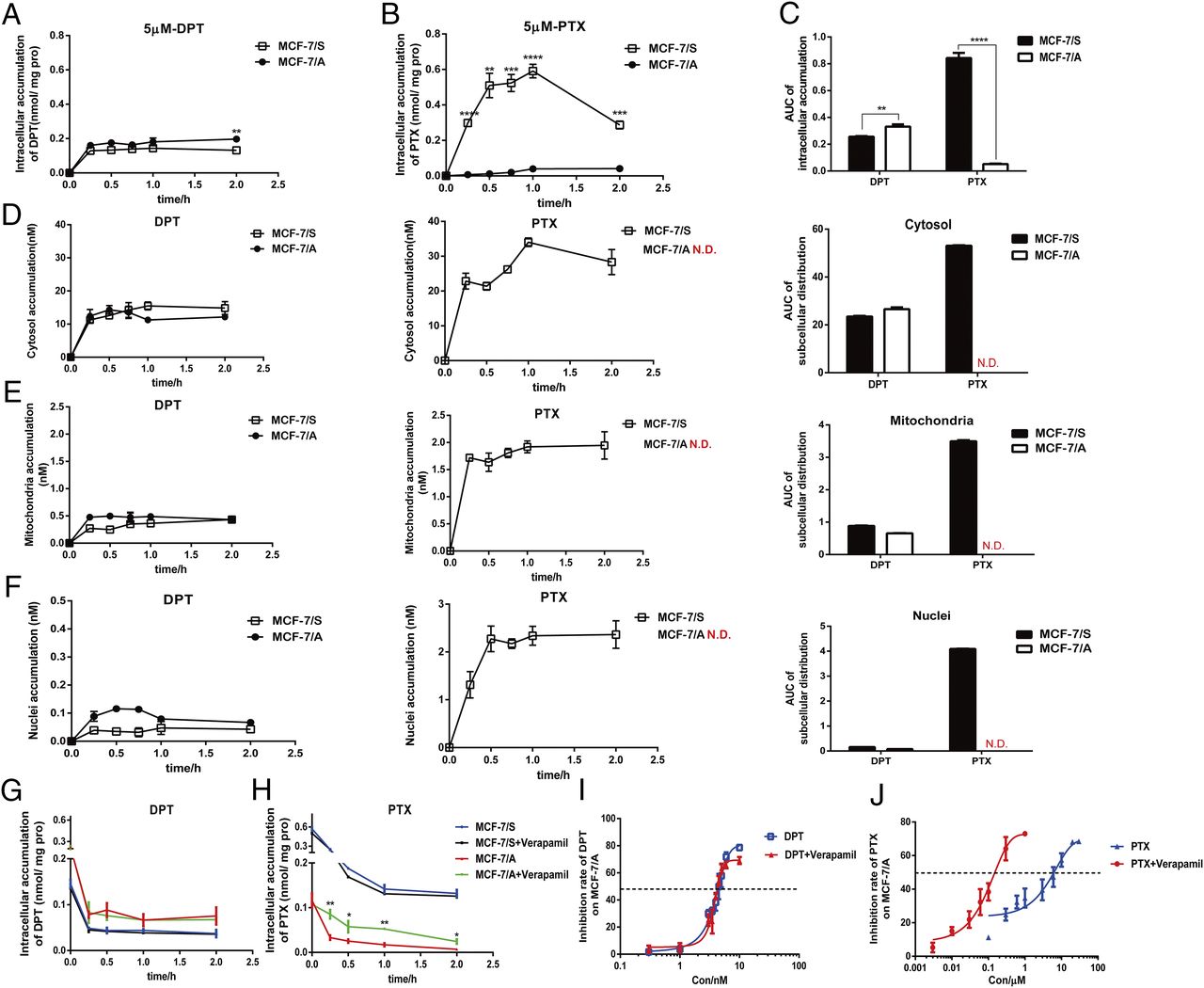
We monitored the intracellular and subcellular concentrations quantitatively by LC/MS/MS to demonstrate further the efficacy of DPT and PTX in MCF-7/S and MCF-7/A cells and to explore the underlying mechanism. As seen in Fig. 6A, the intracellular retention of DPT in MCF-7/A cells was greater than that in MCF-7/S cells during the designated period. Intriguingly, the accumulation of PTX in the MCF-7/A cells was more than 5-fold lower than that in the MCF-7/S cells, which might be the major reason for PTX resistance in the MCF-7/A cells (Fig. 6B). Concurrently and sequentially, we also compared the intracellular accumulation of DPT and PTX in both cell lines at the designated time of 2 hours (Fig. 6C), in line with the preceding results. As the cytosol acted as the target of DPT and PTX, we made further investigations into the subcellular distribution. The time-course analysis revealed the different subcellular distribution of DPT and PTX in both MCF-7/S and MCF-7/A cells (Fig. 6, D–F). As shown in Fig. 6D, both DPT and PTX (89.9% and 92.4%, respectively) were located primarily in the cytosol, but they exerted distinctive retention differences in the MCF-7/S and MCF-7/A cells. These results indicated that the varied effect of DPT and PTX might be due to different intracellular and subcellular accumulations.
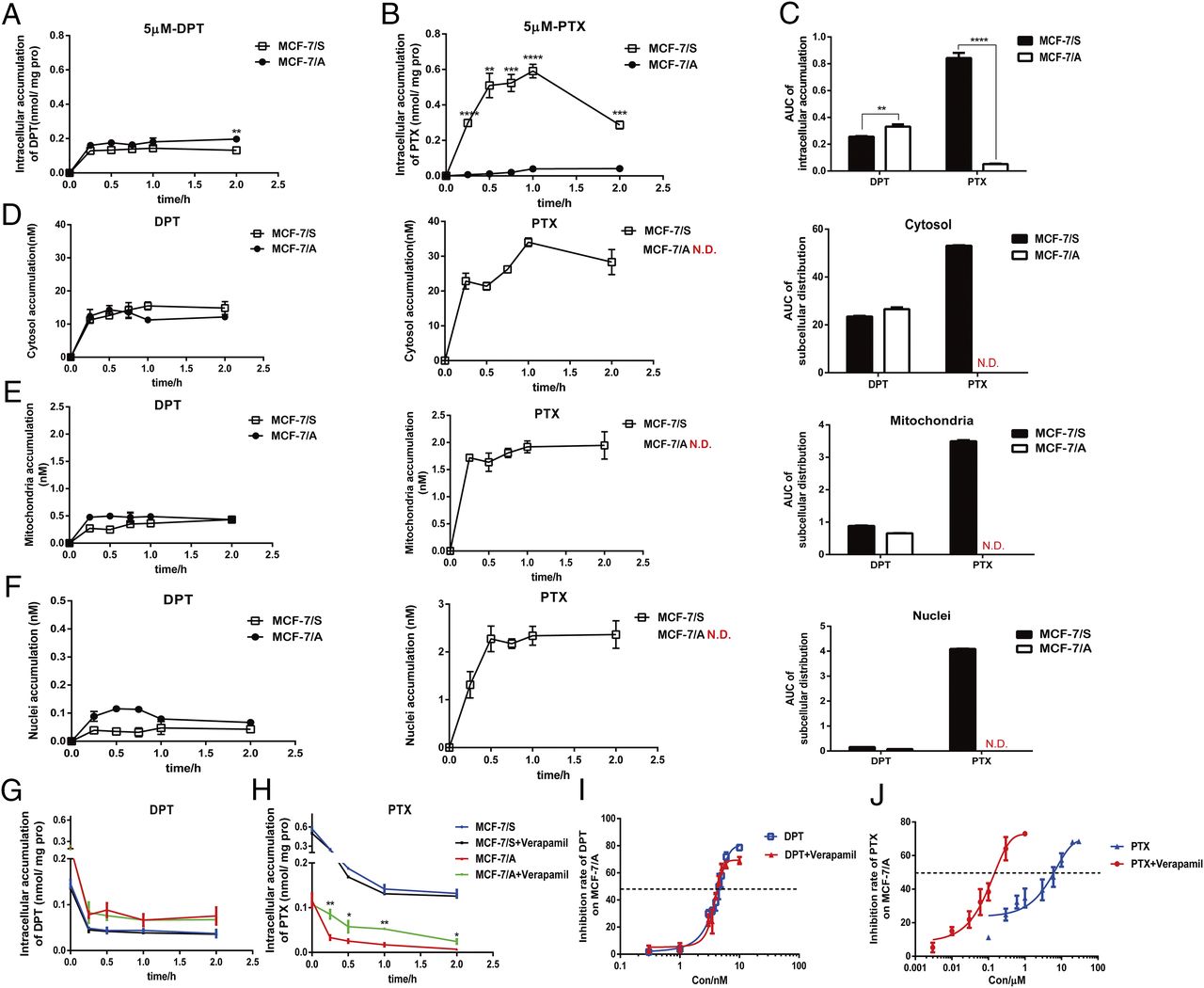
Time-course analysis of intracellular and subcellular accumulation of DPT and PTX in MCF-7/S and MCF-7/A cells. (A and B) Intracellular retention of DPT and PTX in MCF-7/S and MCF-7/A cells. Cells were incubated with 5 µM DPT and PTX for 15, 30, 60, and 120 minutes. (C) Area under the curve of the intracellular accumulation of DPT and PTX in MCF-7/S and MCF-7/A cells at the designated time of 2 hours. (D–F) Time-course analysis and area under the curve of subcellular accumulation of DPT and PTX in the MCF-7/S and MCF-7/A cells. Cells were treated with 1 µM DPT and PTX for 15, 30, and 45 minutes and for 1 and 2 hours. (G and H) Intracellular retention of DPT and PTX in the MCF-7/S and MCF-7/A cells, with or without verapamil. The cells were incubated with 5 µM DPT and PTX for 2 hours, and then the retention of DPT or PTX within cells was monitored with or without verapamil 0, 15, 30 60, and 120 minutes later. (I and J) Cell viability of DPT and PTX in MCF-7/S and MCF-7/A cells with or without verapamil. Results are expressed as mean ± S.D. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. control.
Since P-gp governs the extrude of many anticancer drugs, the efflux assay was also conducted. Intriguingly, the retention curve of DPT in MCF-7/A cells was a little higher than that in MCF-7/S cells, whereas retention of PTX in MCF-7/A cells was decreased to 1 in 15 that in MCF-7/S cells. P-gp inhibitor verapamil did not affect the efflux of DPT either in MCF-7/S or MCF-7/A cells; however, because of the efflux inhibition, accumulation of PTX increased by twice in MCF-7/A cells when pretreated with verapamil (Fig. 6, G and H). Also, PTX exerted greater efficacy in MCF-7/A cells with the verapamil treatment (IC50 = 188.78 ± 26.34 nM) than PTX alone (IC50 = 5011 ± 920 nM), whereas DPT maintained its cytotoxicity in MCF-7/A cells, with (IC50 = 4.47 ± 0.25 nM) or without verapamil (4.64 ± 0.25 nM) (Fig. 6, I and J).
Effect of DPT on Different Efflux Transporters In Vivo and In Vitro.
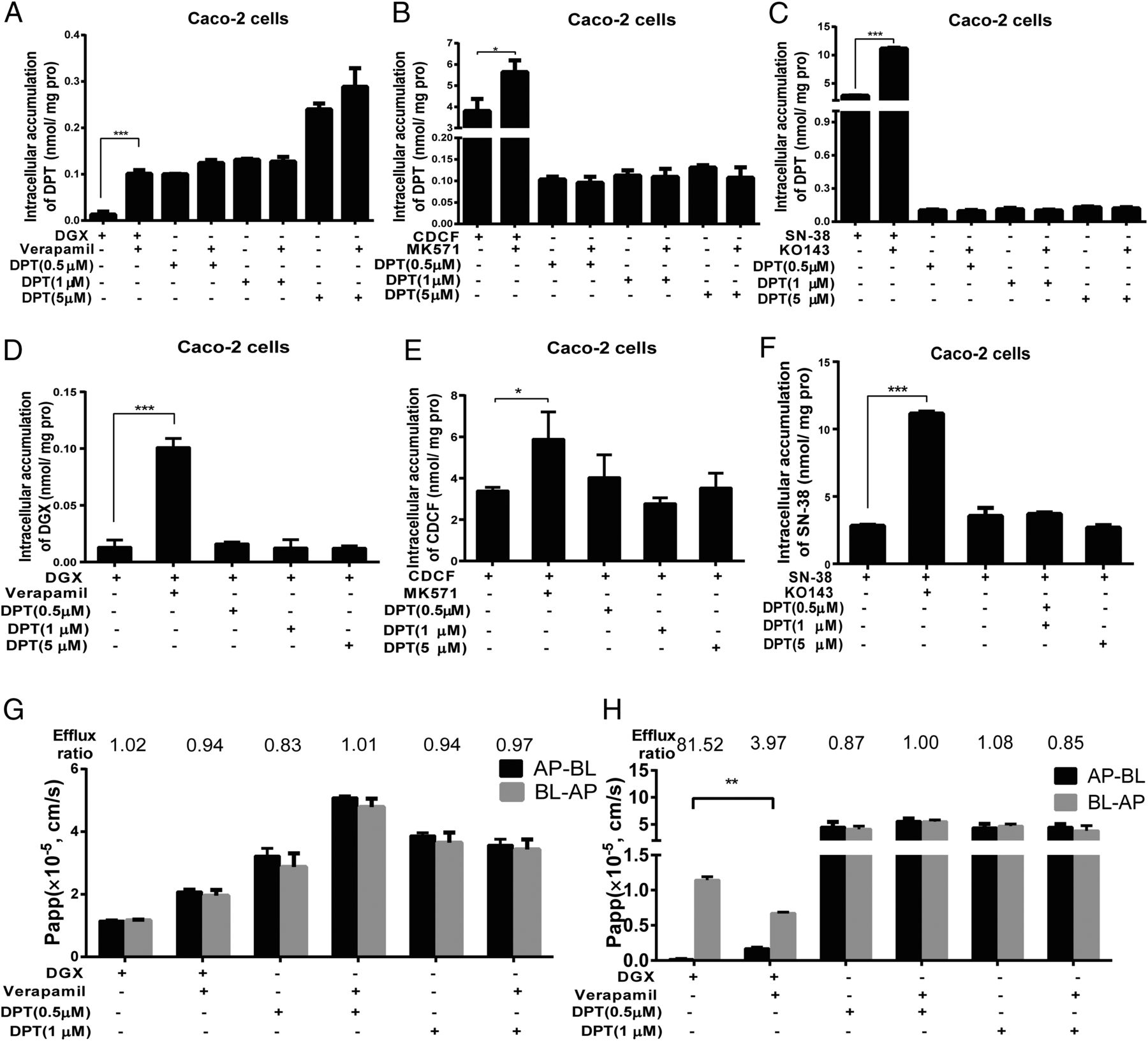
We mined the current available data to further delineate the underlying molecular mechanism for DPT’s better efficacy in the treatment of the MCF-7/A xenografts, and we explored the potential of DPT on different efflux transporters by determining the intracellular accumulation of the classic substrate for rapid screening. As expected, the established P-gp, breast cancer resistance protein (BCRP), and MDR-associated protein 2 (MRP2) inhibitor verapamil (Zhang et al., 2010), KO143 (Matsson et al., 2009), and MK571 (Chen et al., 2017) exhibited a potent inhibitory effect on P-gp, BCRP, and MRP2, respectively, resulting in a palpable increase in the intracellular accumulation of the efflux transporter substrates digoxin (Zhang et al., 2010), SN-38 (Houghton et al., 2004), and CDCF (Chen et al., 2017); however, the retention of DPT did not shift in the absence versus presence of the different inhibitors (Fig. 7, A–C). Subsequently, we ascertained whether DPT could exert an effect on the intracellular accumulation of the substrates. Apparently, DPT exhibited no effect on the retention of the three substrates with various concentrations (Fig. 7, D–F). Combing the results of the preceding intracellular amounts, P-gp, BCRP, and MRP2 might not mediate the transport of DPT.

Effect of DPT on different efflux transporters in vivo and in vitro. (A–C) The accumulation of DPT in Caco-2 cells when treated with different efflux transporter inhibitors. Cells were preincubated with the inhibitors for 1 hour, followed by another 2 hours of incubation in the presence of DPT. The inhibitors investigated (left to right) were verapamil (20 µM), MK571 (10 µM), and KO143 (5 µM). DGX (5 µM), CDCF (10 µM), and SN-38 (10 µM) were used as positive controls. (D–F) The retention of different substrates of the efflux transporters after DPT treatment with different concentrations. Cells were preincubated with DPT for 1 hour, followed by another 2 hours of incubation in the presence of DGX, CDCF, and SN-38. (G and H) Effect of DPT on the transport of P-gp substrates across the WT-MDCK and MDR1-MDCK cell monolayers. DPT (0.5 and 1 µM) was loaded on either the anteroposterior (AP) or basolateral (BL) side and incubated for 2 hours. DGX was treated as a P-gp substrate control. Verapamil was used as a P-gp inhibitor control. Data are presented as mean ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus control. AP, apical; BL, basolateral.
To confirm more completely that DPT was not the substrate of P-gp, we monitored the flux of DPT across WT-MDCK and MDR1/P-gp overexpressing MDR1-MDCK cell monolayers in the AP-BL and BL-AP directions in the absence or presence of verapamil (Fig. 7, G and H). Wild-type MDCK cells were applied as a negative control with a low expression of constitutive canine P-gp. Consistent with the results of the Caco-2 retention studies, the ER of DPT was approximately 1.0, which was less than the threshold of 2.0 for defining whether the drug acted as the substrate of P-gp or not and did not change in the absence or presence of verapamil. In contrast, verapamil remarkably increased the transport of DGX in the anteroposterior-basolateral direction and decreased in the basolateral-anteroposterior direction, with the ER changing from 81.52, which was far more than 2.0 to 3.97. Thus, DPT was not the substrate of P-gp.
Discussion
The incidence and mortality of breast cancer have increased throughout the world (Althuis et al., 2005), and it remains the most common cancer in women (Murray et al., 2012). In recent years, more and more drugs explored in clinical trials have provided promising advances in breast cancer treatments. Microtubule-binding agents are among the efficacious chemotherapeutic drugs that are commonly used for the treatment of breast cancer. Although antimicrotubule drugs, such as PTX and docetaxel, have been successfully applied in the clinic, drug resistance often occurred (Kavallaris, 2010) and acted as an obstacle for first-line chemotherapy. Augmented efflux transporters and diverse β-tubulin isotypes were involved in the development of drug resistance (Dumontet and Sikic, 1999). Thus, there is a pressing need to develop new microtubule inhibitors that can escape from the efflux of transporters related to MDR.
As a promising microtubule inhibitor candidate, DPT possesses various pharmacologic activities, including anti-inflammatory (Jin et al., 2008), antiviral (Gordaliza et al., 1994), antiangiogenic (Wang et al., 2015a), and antitumor effects, among which antitumor attracted the most attention. Previous studies have shown that DPT inhibits the proliferation of SGC-7901 cancer cells and induces G2/M cell-cycle arrest (Wang et al., 2015b). Animal experiments revealed that DPT showed potent toxicity in lung cancer (Wu et al., 2013). Additionally, glioma growth could be inhibited by DPT in vivo and in vitro, which could be attributed to the upregulation of PARP-1 and AIF nuclear translocation (Ma et al., 2016). Although DPT induced apoptosis of the human BC mDA-MB-231 cell line in vitro (Benzina et al., 2015), little research has been conducted on the efficacy of DPT on the MCF-7 cell line. Whether DPT avoids MDR has yet to be investigated. In our study, Adriamycin-resistant MCF-7/A cells were used to explore the influence of DPT on MDR . It is noteworthy that the cytotoxicity of DPT in drug-resistant MCF-7/A cells equals that in drug-sensitive MCF-7/S cells, as indicated by IC50, which is much better than the effect of PTX. In current studies, several lines of evidence indicated that DPT exhibited an inhibitory effect on the growth of various tumors. In a concentration-dependent manner, DPT exerted antitumor effects better than etoposide on H460 xenografts but with fewer side effects (Wu et al., 2013). Additionally, strong tumor inhibition properties were also observed on MDA-MB-231 human BC xenografts in the BALB/c nude mice model (Khaled et al., 2016). According to previous studies, we chose 12.5 mg/kg of DPT formulated as β-cyclodextrin inclusion complex (Zhu et al., 2010) as the target dose for intravenous injection to illuminate the in vivo antitumor effect of DPT with the MCF-7/S and MCF-7/A xenograft models. On the MCF-7/S xenograft models, DPT showed comparable inhibition effects to PTX as depicted in Fig. 2. Surprisingly, DPT maintained its tumor growth suppression of the MCF-7/A xenograft models, whereas PTX exerted a marginal inhibitory rate, suggesting that PTX had developed resistance. This was further evidenced by the histopathological examination of the tumors. The concentration of DPT in the MCF-7/A xenografts was greater than that in the MCF-7/S xenografts, along with the plasma/tumor ratio. To our knowledge, this was the first time the proliferation inhibition of DPT on MCF-7/S cells and related drug-resistant MCF-7/A cells in vivo and in vitro was proven.
Since DPT exerted a similar cytotoxicity on both the drug-sensitive and drug-resistant xenograft models in vivo, further in vitro data confirmed the equal cytotoxicity of DPT on drug-sensitive and resistant MCF-7 cells with the indicated cell-cycle assay. These results revealed that DPT might overcome MDR in comparison with PTX. Although they are both microtubule-targeting agents, DPT may act in a different mode from PTX. Tubulin-targeting agents are generally classified into two groups: microtubule-stabilizing agents, such as paclitaxel, and microtubule-destabilizing agents, such as colchicine. PTX was investigated to reduce the purified tubulin subunits that are critical for polymerization into microtubules (Weaver, 2014) and to shift the tubulin from its assembly equilibrium to its polymeric state (Prota et al., 2013). The results of the microtubule polymerization test confirmed the different binding modes of DPT and PTX. How DPT acts on microtubules may be the cause of its escape from the resistance transporters. Therefore, this discrepancy underscores the need to determine the underlying mechanism.
From the cellular pharmacokinetics perspective (Zhou et al., 2011), after penetrating the cell membrane, the anticancer drugs must bind to the intracellular target, thus exerting efficacy. Therefore, the intracellular distribution and disposition processes of the drugs acted as the determinants of their therapeutic effect (Krishan et al., 1997; Duvvuri and Krise, 2005). As reported, the weakened efficacy of PTX was ascribed to the efflux transporters, which not only restricted the distribution of the drug intracellularly, but they also reduced intracellular exposure (Argov et al., 2010). Here, we established a novel LC/MS/MS method to compare the intracellular accumulation of PTX and DPT. From our results, the time-concentration curve demonstrated remarkable differences in the intracellular behavior of PTX between the MCF-7/S and MCF-7/A cells, whereas DPT exerted a similar retention in the two cell lines (Fig. 6). This finding is in line with our previous data revealing the discrepancy involving the accumulation of DPT and PTX in the MCF-7/S and MCF-7/A xenografts. Support for this notion came from our findings that the subcellular distribution of DPT and PTX exhibited distinctive differences in the accumulation in different cellular organs, especially in cytosol. Based on the efflux assay, all the results indicated that the different behaviors of intracellular uptake, even subcellular disposition in drug-sensitive and drug-resistant cells, might determine their therapeutic efficacy (Xiong et al., 2010).
Then, Caco-2 cells and MDR1-MDCK cells were used to verify that DPT was not a substrate of these efflux transporters related to MDR. Because of the rich expression of the efflux transporters in the apical membrane, Caco-2 cell monolayers have been widely used to study drug intestinal absorption and the possible interaction with efflux transporters (Zhang et al., 2010). We provided ample evidence that DPT might not be the substrate of the three efflux transporters (P-gp, BCRP, and MRP2). In emerging studies, the MDR1-MDCK cell model has been used to screen P-gp substrates or inhibitors, expressed by apparent permeability (Papp). We found that the ER of DPT did not change with or without verapamil, and it was less than the threshold for the definition of a P-gp substrate, which confirmed that DPT was not the substrate of P-gp. The findings may illustrate DPT’s better efficacy in the treatment of MCF-7/A xenografts.
In consideration of all these findings, we have provided several lines of evidence that first illuminate that DPT exerted a proliferation inhibition effect on MCF-7/S and resistant MCF-7/A cells in vitro and in vivo in comparison with PTX. Moreover, we initially confirmed that DPT was not a substrate of the P-gp efflux pump and could overcome P-gp-mediated MDR. Thus, DPT, as a new tubulin polymerization inhibitor, can be exploited as a promising agent for the treatment of MDR tumors.
Acknowledgments
The authors thank all the staff, colleagues, and students during the animal experiments and to LetPub (www.letpub.com) for its linguistic assistance during revision of this manuscript.
Authorship Contributions
Participated in research design: Zang, Wang, Hao, Zhou.
Conducted experiments: Zang, Cai, Zhang, Chen, Hao, Zhou.
Contributed new reagents or analytical tools: Zang, Cai, Wu, Zhu, Zhou.
Performed data analysis: Zang, Cai, Zheng, Chen, Wu, Zhou.
Wrote or contributed to the writing of the manuscript: Zang, Wang, Hao, Zhou.
Footnotes
- Received November 10, 2017.
- Accepted February 22, 2018.
↵1 X.Z. and G.W. contributed equally to this work.
The work was supported by the China National Nature Science Foundation [Grants 81573496 and 81530098], Jiangsu Province Key Laboratory of Drug Metabolism and Pharmacokinetics Projects [Grant BM2012012], Jiangsu Province Nature Science Foundation [Grant BK20160076], China “Creation of New Drugs” Key Technology Projects [Grant 2015ZX09501001], the Foundation for Innovative Research Groups of the National Natural Science Foundation of China [Grant 81421005], and the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University [Grant SKLNMZZCX201608].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BCRP
- breast cancer resistance protein
- DPT
- deoxypodophyllotoxin
- ER
- efflux ratio
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MCF-7/A
- MCF-7/Adr
- MDCK
- Madin–Darby canine kidney epithelial cells
- MDR
- multidrug resistance
- MRP2
- multidrug resistance-associated protein 2
- Papp
- apparent permeability values
- P-gp
- P-glycoprotein
- PTX
- paclitaxel
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}