


Abstract
Vitamin D3 is an important prohormone critical for maintaining calcium and phosphate homeostasis in the body and regulating drug-metabolizing enzymes and transporters. 25-Hydroxyvitamin D3 (25OHD3), the most abundant circulating metabolite of vitamin D3, is further transformed to the biologically active metabolite 1α,25-dihydroxyvitamin D3 (1α,25-(OH)2D3) by CYP27B1 in the kidney and extrarenal tissues, and to nonactive metabolites by other cytochrome P450 enzymes. In addition, 25OHD3 undergoes sulfation and glucuronidation in the liver, forming two major conjugative metabolites, 25OHD3-3-O-sulfate (25OHD3-S) and 25OHD3-3-O-glucuronide (25OHD3-G), both of which were detected in human blood and bile. Considering that the conjugates excreted into the bile may be circulated to and reabsorbed from the intestinal lumen, deconjugated to 25OHD3, and then converted to 1α,25-(OH)2D3, exerting local intestinal cellular effects, it is crucial to characterize enterohepatic transport mechanisms of 25OHD3-S and 25OHD3-G, and thereby understand and predict mechanisms of interindividual variability in mineral homeostasis. In the present study, with plasma membrane vesicle and cell-based transport studies, we showed that 25OHD3-G is a substrate of multidrug resistance proteins 2 and 3, OATP1B1, and OATP1B3, and that 25OHD3-S is probably a substrate of breast cancer resistance protein, OATP2B1, and OATP1B3. We also demonstrated sinusoidal and canalicular efflux of both conjugates using sandwich-cultured human hepatocytes. Given substantial expression of these transporters in liver hepatocytes and intestinal enterocytes, this study demonstrates for the first time that transporters could play important roles in the enterohepatic circulation of 25OHD3 conjugates, providing an alternative pathway of 25OHD3 delivery to the intestinal tract, which could be critical for vitamin D receptor-dependent gene regulation in enterocytes.
Introduction
Vitamin D3 is essential for regulation of calcium and phosphate homeostasis in the body. Vitamin D3 exerts most of its biologic functions through a biologically active metabolite, 1α,25-dihydroxyvitamin D3 (1α,25-(OH)2D3), which binds with high affinity to vitamin D receptor (VDR) and activates transcription of target genes (Christakos, 2012). Besides regulating intestinal absorption of calcium and phosphate, VDR signaling also appears to be important for regulation of human intestinal CYP3A4 expression (Thummel et al., 2001), which has been shown to be associated with seasonal changes in vitamin D production (Thirumaran et al., 2012) and can be modified by vitamin D supplementation (Schwartz, 2009).
The biologically active metabolite 1α,25-(OH)2D3 is generated following two metabolic steps (Fig. 1). First, 25-hydroxyvitamin D3 (25OHD3) is produced from vitamin D3 primarily by hepatic CYP2R1. 25OHD3, which is the major circulating form of vitamin D3 (Henry and Norman, 1984), tightly binds to plasma proteins (e.g., vitamin D-binding protein, DBP) and exhibits a relatively long (∼14 days) plasma half-life, and therefore is used as a biomarker for vitamin D exposure (Holick, 2007). 25OHD3 is then further metabolized to 1α,25-(OH)2D3 primarily by CYP27B1 in the kidney and certain extrarenal tissues (Hewison et al., 2007; Feldman et al., 2014).

Major metabolic pathways of vitamin D3 in humans.
Although the kidney is generally regarded to be the main source of 1α,25-(OH)2D3, CYP27B1 is also expressed in a number of extrarenal tissues, including intestinal epithelial cells (Bikle, 2009, 2014). Importantly, as suggested by Balesaria et al. (2009) and others (Bises et al., 2004), 1α,25-(OH)2D3-mediated biologic effects in the intestine might also be produced via an intracrine process involving the bioactivation of 25OHD3 by intestinal CYP27B1. A recent study provided direct experimental evidence that CYP27B1 in duodenum indeed can metabolize 25OHD3 and contribute to local 1α,25-(OH)2D3 production in the intestine (Gawlik et al., 2015). However, the delivery of 25OHD3 to the intestinal mucosal cells remains unclear but could involve production and delivery of vitamin D3 conjugates.
Studies in humans using radioactive compounds suggested that vitamin D3 and its hydroxylated metabolites are excreted through the bile in the forms of conjugates (Avioli et al., 1967). In one study, it was suggested that 25OHD3 undergoes extensive enterohepatic circulation, where 85% of secreted radioactivity into duodenum was reabsorbed in 24 hours after intravenous administration of [3H]-25OHD3 (Arnaud et al., 1975). Considering the wide distribution of β-glucuronidase (Whiting et al., 1993; Oleson and Court, 2008) and steroid sulfatase (Miki et al., 2002) in intestinal bacteria and within the enterocyte, conjugated 25OHD3 in the bile may be reabsorbed from the intestinal lumen in an intact form or as an aglycone after hydrolysis by enzymes or bacteria in the intestinal tract.
In addition to 1α-hydroxylation, 25OHD3 undergoes a series of catabolic hydroxylation reactions (e.g., 23S, 24R, and 4β) that mediate inactivation of vitamin D3 and its excretion (Christakos et al., 2010; Wang et al., 2012). 25OHD3 also undergoes sulfation and glucuronidation in the liver, generating two major circulating conjugative metabolites, 25OHD3-3-O-sulfate (25OHD3-S) (Wong et al., 2018) and 25OHD3-3-O-glucuronide (25OHD3-G) (Wang et al., 2014) (Fig. 1). In healthy humans, plasma concentrations can reach as high as 185 nM for 25OHD3, 100 nM for 25OHD3-S, and 10 nM for 25OHD3-G (Gao et al., 2017). Both conjugative metabolites are tightly bound to DBP, with an affinity similar to that of 25OHD3, and were not detected in the urine of healthy individuals. The high protein binding of vitamin D3 conjugates in the blood not only reduces urinary excretion from the body but also greatly limits the passive diffusion and active transport across membrane barriers and thus minimizes the delivery of unbound vitamin D-derived conjugates to target tissues (e.g., intestinal epithelia) in the body. In contrast, owing to the absence of binding proteins in the bile, biliary conjugates of vitamin D3 should be directly accessible to the intestinal epithelia cells and uptake transporters for hormone conjugates (Hofmann, 2011). We hypothesized that 25OHD3 conjugates excreted into the bile are deconjugated by bacterial or intracellular sulfatase and glucuronidase and then hydroxylated by intestinal CYP27B1 to 1α,25-(OH)2D3, thereby regulating VDR target gene expression.
To test the hypothesis that the regulation of intestinal VDR target genes is mediated in part by 1α,25-(OH)2D3 in the intestinal tract, it is essential to understand the hepatic and intestinal transport of conjugative 25OHD3 metabolites. However, to date, there is essentially nothing known about these processes. The objective of the current study was to characterize the major uptake and efflux transport mechanisms for 25OHD3-S and 25OHD3-G in the liver.
Materials and Methods
Materials.
Ko143, MK571, indomethacin, rifampin, cyclosporine A (CsA), fumitremorgin C, cholic acid, taurocholate, estrone-3-sulfate (E1-3-S), estradiol-17β-glucuronide (E2-17β-G), estradiol-3-sulfate (E2-3-S), 3′-phosphoadenosine-5′-phosphosulfate, ATP disodium salt, adenosine 5′-monophosphate (AMP) monohydrate, and glutathione were purchased from Sigma-Aldrich (St. Louis, MO). 25OHD3, d6-25OHD3, 25OHD3-S, and 25OHD3-G were obtained from Toronto Research Chemicals (Toronto, Canada). Hygromycin B, G-418, and RIPA lysis buffer were purchased from (Thermo Fisher Scientific, Waltham, MA). All other buffers and chemicals were of the highest grade commercially available.
Cell culture medium, antibiotic-antimycotic solution (100× ) and Hanks’ balanced salt solution (HBSS) were purchased from Invitrogen/Thermo Fisher Scientific (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from VWR (Radnor, PA). Plasma membrane vesicles, including Sf9 cells overexpressing breast cancer resistance protein (BCRP), MRP2, or MRP3; HEK293 cell line cells overexpressing MRP4; and their corresponding control membrane vesicles were purchased from SOLVO Biotechnology (Szeged, Hungary). Flp-In-Chinese hamster ovary (CHO)/OATP2B1, Flip-In-CHO mock, CHO/OATP1B1, CHO/OATP1B3, and wild-type CHO cells were provided by Dr. Bruno Stieger of the Department of Clinical Pharmacology and Toxicity, University Hospital Zurich, Switzerland, through Dr. Bruno Hagenbuch of the Department of Pharmacology, Toxicology, and Therapeutics, University of Kansas Medical Center. Preplated sandwich-cultured human hepatocytes (SCHHs) were obtained from In Vitro ADMET Laboratories, LLC (IVAL; Malden, MA). Multiscreen HTS Vacuum Manifold and 96-well filter plates with glass fiber filters were purchased from Merck Millipore (Billerica, MA). Ultracentrifuge tubes (Beckman 343775) for unbound fraction determination were purchased from Beckman Coulter (Brea, CA). Anonymous, pooled human bile was kindly provided by Dr. Evan D. Kharasch at the Washington University in St. Louis (St. Louis, MO).
Human Bile Analysis.
Pooled human bile from different donors was analyzed with a sensitive liquid chromatography–tandem mass spectrometry (LC-MS/MS) method coupled with derivatization (Gao et al., 2017), and deuterated internal standards (d6-25OHD3-S and d6-25OHD3-G) used in the analysis were obtained through biotransformation from d6-25OHD3 as previously described (Gao et al., 2012b). In brief, human bile (100 μl) was spiked with d6-25OHD3-S and d6-25OHD3-G and was then precipitated with 200 μl acetonitrile. The resulting supernatant was buffered with 1 ml of 0.1 M sodium acetate (pH 3.2) and subjected to solid-phase extraction (SPE) using Waters Oasis WAX (60 mg, 3 cc) anion exchange cartridges. Eluates from the SPE columns were then dried and reconstituted in 10 μl of methanol and derivatized with 4-(4′-dimethylaminophenyl)-1,2,4-triazoline-3,5-dione (DAPTAD) for 1 hour at room temperature in the dark. The reaction mixture was evaporated under N2 flow and reconstituted in 100 μl of mobile phase. A centrifugation step (13,362g for 5 minutes) was applied to remove insoluble materials prior to LC-MS/MS analysis. DAPTAD-25OHD3-S, DAPTAD-d6-25OHD3-S, DAPTAD-25OHD3-G, and DAPTAD-d6-25OHD3-G were analyzed using an AB Sciex QTRAP 6500 LC-MS/MS system. Owing to the lack of blank bile matrix, calibration curves were plotted with corrected peak area ratios of DAPTAD-25OHD3-S/G and DAPTAD-d6-25OHD3-S/G by subtracting “blank” samples from those spiked with serial 25OHD3-S and 25OHD3-G solutions.
Inside-Out Membrane Vesicular Uptake Assay.
A rapid filtration technique modified from a previously reported method (Gao et al., 2012a) was used for inside-out plasma membrane vesicular transport assays. In brief, plasma membrane vesicles (25 μg protein) and test compounds of various concentrations were incubated at 37°C in the presence of ATP or AMP (5 mM) in a buffer (pH 7.0) containing 10 mM Tris/HCl, 10 mM MgCl2, and 250 mM sucrose for HEK293 membrane vesicles or in a buffer (pH 7.0) containing 40 mM MOPS/Tris, 70 mM KCl, and 7.5 mM MgCl2 for Sf9 plasma membrane vesicles. Glutathione (2 mM) was added to the incubations for multidrug resistance protein (MRP) transporters. Transport was terminated after incubation for a designated time by the addition of 200 μl of ice-cold buffer [containing 1.5% bovine serum albumin (BSA) for 25OHD3-S and 25OHD3-G]. The mixture was rapidly transferred to a 96-well glass fiber filter plate and then washed five times with 200 μl of ice-cold wash buffer (containing 1.5% BSA for 25OHD3-S and 25OHD3-G). The compound trapped in membrane vesicles was retained on the filters and eluted by the addition of 200 μl of methanol containing the corresponding internal standard. E2-17β-G, a known substrate of MRPs and BCRP, was used as a positive control for MRPs and BCRP or a probe substrate of these transporters. In inhibitory studies using E2-17β-G as a probe substrate, E2-17β-G at 10 μM (for BCRP and MRP4) or 2 μM (for MRP2 and MRP3) were incubated with transporter-overexpressing membrane vesicles (25 μg) in the presence of varying concentrations of 25OHD3-S. The incubations were conducted and terminated under the same conditions as described above. Vehicle (dimethyl sulfoxide) used to dissolve test compounds was kept below 0.2% (v/v) in all the incubations and no effects of the vehicle at concentrations below 0.2% (v/v) on vesicular uptake or nonspecific binding of 25OHD3-S and 25OHD3-G were observed. ATP-dependent uptake of 25OHD3-S, 25OHD3-G, and E2-17β-G into inside-out plasma membrane vesicles was calculated by subtracting the uptake in the presence of AMP from that in the presence of ATP.
Unbound Fractions of 25OHD3-S and 25OHD3-G in Plasma Membrane Vesicle Incubations.
Ultracentrifugation was used to determine the unbound fraction of 25OHD3-S and 25OHD3-G in incubation buffers of membrane vesicular transport assays as previously described (Shirasaka et al., 2013). Briefly, 25OHD3-S or 25OHD3-G at various concentrations was incubated with mock Sf9 or HEK293 membrane vesicles in ultracentrifuge tubes (Beckman 343775) under the same experimental conditions as the vesicular transport assays (e.g., temperature and incubation time). After incubation, the mixture was centrifuged at 435,000g at 37°C for 90 minutes using a Sorvall Discovery M150 SE ultracentrifuge (Thermo Fisher Scientific) and a Thermo Fisher Scientific S100-AT3 rotor. The unbound fraction (%) was calculated by dividing the concentration of 25OHD3-S or 25OHD3-G in the supernatant after centrifugation by the total concentration in a parallel mixture incubated at 37°C for 90 minutes without centrifugation.
Culture of Organic Anion Transporting Polypeptide–Overexpressing Cells.
Cells were maintained at 37°C in a humidified 5% CO2 incubator. Flp-In-CHO cells transfected with OATP2B1 (Flp-In-CHO/OATP2B1) and mock control cells were cultured as previously described (Pacyniak et al., 2010). In brief, cells were maintained in F-12 Nutrient Mixture (Gibco, Thermo Fisher Scientific Inc., Waltham, MA) containing 10% fetal bovine serum, 2 mM l-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, and 400 μg/ml hygromycin B. CHO cells stably expressing OATP1B1 (CHO/OATP1B1) and OATP1B3 (CHO/OATP1B3) were cultured in phenol red-free, low-glucose DMEM media with 10% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, and 50 μg/ml l-proline in the presence of 500 μg/ml G418 as previously described (Gui et al., 2010). Wild-type CHO parent cells were cultured in the same media as above without G418.
Cellular Uptake Transport Assay.
OATP-transfected CHO cells and respective control cells were seeded on CELLCOAT 24-well plates (Greiner Bio-One, Monroe, NC) at a density of approximately 4 × 104 cells/well. For CHO/OATP1B1, CHO/OATP1B3, and wild-type CHO parent control cells, after cells were plated and cultured for 24 hours, 5 mM Na-butyrate was added to culture medium and the culture was continued for another 24 hours to induce OATP expression. Flp-In-CHO/OATP2B1 and mock control CHO cells were cultured in media without Na-butyrate for 48 hours straight. Prior to uptake experiments, cells were washed and equilibrated in HBSS at 37°C for 10 minutes. Uptake was initiated by addition of 0.5 ml of HBSS containing 25OHD3-S or 25OHD3-G at various concentrations to corresponding cells and terminated after incubation for 5 minutes at 37°C by aspiration of incubation buffer and washing of the cells twice with 2 ml of ice-cold fresh HBSS (containing 1.5% BSA for 25OHD3-S and 25OHD3-G). An aliquot of 50 μl of the uptake incubation buffer was collected for each concentration to determine free concentrations of 25OHD3-S or 25OHD3-G in the incubation buffers. After the uptake, cells were lysed with 0.25 ml of methanol containing corresponding internal standard and centrifuged at 13,362g for 5 minutes. The supernatant was then evaporated and reconstituted in 100 μl of mobile phase for LC-MS analysis. In parallel, cells grown under the same conditions were lysed with 0.25 ml of RIPA cell lysis buffer and protein concentrations of cell lysates were determined using Pierce BCA protein assay kit (Pierce Chemical, Rockford, IL). Accumulation (or uptake) of 25OHD3-S or 25OHD3-G in cells was normalized to protein content of the cells. For positive control, transport of E2-17β-G (OATP1B1 and OATP1B3) and E1-3-S (OATP2B1) was conducted at 10 μM under the same conditions as described above.
Data Analysis for Uptake Assays.
All the membrane vesicular and cellular uptake experiments were performed in triplicate or quadruplicate with at least two independent repeats. Differences in membrane vesicular uptake between ATM and AMP or in cellular accumulation between organic anion transporting polypeptide (OATP)–expressing and control cells were analyzed using unpaired Student’s t test. Differences with P value of <0.05 were considered statistically significant. Km and Vmax of transport kinetics were estimated by fitting of experimental data to the Michaelis-Menten equation with nonlinear regression. IC50 values were also determined by nonlinear regression as previously described (Duan et al., 2015). All the analyses were performed using the GraphPad Prism software (vers. 5.01; La Jolla, CA).
Efflux of 25OHD3-S or 25OHD3-G from Sandwich-Cultured Human Hepatocytes.
SCHHs were obtained from five female donors (HH1055, HH1085, HH1007, and HH1103) and one male donor (HH1031). SCHHs were plated in 96-well plates on day 1 by the vendor and shipped with 37°C heating patches overnight on day 2. Upon arrival of cells, the culture media was replaced with fresh and warm hepatocyte maintenance media provided by the vendor and the cells were cultured for another 2 days with medium change every day. On day 5, cells were washed and preincubated in HBSS with 25OHD3 at 10 μM for 4 hours and efflux was initiated by washing and incubating cells with 100 μl of fresh HBSS buffer with or without calcium. Human vitamin D-binding protein was added to incubation buffer to 3 μM concentration during the efflux phase to reduce nonspecific binding of 25OHD3 conjugates (e.g., to cell membrane and plastic cell culture flask). To confirm if efflux transporters are involved in the canalicular and sinusoidal efflux of 25OHD3-S and 25OHD3-G, the BCRP inhibitor fumitremorgin C (FTC) (10 μM) or the MRP inhibitor MK571 (40 μM) was added to the incubation buffer throughout the 4-hour loading and 20-minute efflux phases. As a positive control, SCHHs were loaded with 2 μM taurocholate for 20 minutes, and the efflux was initiated by washing and incubating the cells with fresh HBSS with or without calcium. During the efflux phase, incubation buffer (100 μl) was collected at designated time points from corresponding wells that were loaded with 25OHD3 or taurocholate. The cells were lysed with 100 μl methanol and collected at the end of the efflux phase. For 25OHD3 group, collected incubation buffers and cell lysates were spiked with deuterated internal standards d6-25OHD3-S and d6-25OHD3-G and evaporated to dryness before being derivatized with DAPTAD. For the taurocholate group, incubation buffers were spiked with internal standard cholic acid and evaporated to dryness before being reconstituted in 100 μl of mobile phase. The resulting samples were subject to quantification by LC-MS/MS as described below.
In the presence of calcium, the tight junctions between hepatocytes and the canalicular networks remain intact, allowing measurement of efflux across the sinusoidal membrane of hepatocytes. Removal of calcium from the incubation buffer disrupts the tight junctions and allows measurement of efflux across both the canalicular and sinusoidal membranes. Therefore, the difference in efflux between the with- and without-calcium samples represents biliary efflux of a test compound. Biliary efflux index (BEI) was calculated using the equation: BEI (%) = [Efflux (sinusoidal + canalicular) − Efflux (sinusoidal)]/Efflux (sinusoidal + canalicular)] × 100.
Analytical Methods.
Given the simple matrix and relatively high concentrations, 25OHD3-S and 25OHD3-G in vesicular and cellular uptake, as well as the unbound fraction-determination assays, were quantified using a straightforward LC-MS method reported by Wang et al. (2014). As for experiments with SCHHs, owing to low production of 25OHD3-S and 25OHD3-G in hepatocytes, 25OHD3-S and 25OHD3-G in the incubation buffers and cell lysates were derivatized with DAPTAD and quantified using a sensitive LC-MS/MS method as previously described (Gao et al., 2017).
E2-17β-G in the membrane vesicular and cellular uptake assays was quantified using LC-MS with E2-3-S as the internal standard. Briefly, chromatographic separation was achieved on a Waters Symmetry C18 (2.1 × 50 mm, 3.5 μm) column (Waters, Milford, MA) with a mobile phase consisting of 1 mM ammonium chloride (A) and methanol (B) at 45°C. A linear gradient from 20% B (0–0.5 minutes) to 75% B (4 to 5 minutes) in 3.5 minutes at a 0.25 ml/min flow rate was employed. The mass spectrometer was operated in the negative ionization mode. E2-17β-G and E2-3-S were detected by selective ion monitoring at m/z 447 and m/z 351, respectively.
E1-3-S in the OATP2B1-overexpressing cell uptake assay was quantified using LC-MS with E2-3-S as the internal standard. Chromatographic separation was achieved using a Waters Symmetry C18 (2.1 × 50 mm, 3.5 μm) column on an Agilent 1200 LC system. The elution was performed at a flow rate of 0.25 ml/min with the mobile phase containing 0.1% formic acid in water and methanol at a ratio of 30:70 (v/v). Mass spectrometric analysis was carried out using a negative mode electrospray ionization method on an Agilent 6410 triple quadrupole tandem mass spectrometer. Single-ion monitoring at m/z 351 and m/z 349 was applied for detection of E1-3-S and E2-3-S, respectively.
Taurocholate in SCHH efflux experiments was quantified using cholic acid as the internal standard. Chromatographic separation was achieved using a Waters Symmetry C18 (2.1 × 50 mm, 3.5 μm) column on an Agilent 1200 LC system. The elution was performed at a flow rate of 0.25 ml/min with the mobile phase containing acetonitrile and 10 mM ammonium acetate (native pH) at a ratio of 64:36 (v/v). Mass spectrometric analysis was carried out using a negative mode electrospray ionization method on an Agilent 6410 triple quadrupole tandem mass spectrometer. Single-ion monitoring at m/z 514 and m/z 407 was applied for detection of taurocholate and cholic acid, respectively.
The LC-MS quantification methods for E2-17β-G, E1-3-S and taurocholate were developed in-house; a linear standard curve (r2 > 0.99) was established for each analytical batch, and the accuracy of interday and intraday quality control samples was within the range of 85%–115% during the analysis.
Results
Human Bile Analysis.
To confirm the presence of 25OHD3-S and 25OHD3-G in human bile, pooled healthy human bile was derivatized with DAPTAD and analyzed using a sensitive LC-MS/MS method we developed (Gao et al., 2017). As shown in Fig. 2, peaks were observed in bile for both DAPTAD-25OHD3-S (Fig. 2A) and DAPTAD-25OHD3-G (Fig. 2C) at the expected slightly longer retention times shown by their corresponding deuterated internal standards (Fig. 2, B and D). On the basis of the calibration curves established with 25OHD3-S and 25OHD3-G standards, concentrations of 25OHD3-S and 25OHD3-G in the pooled human bile were estimated to be around 2.55 and 0.30 nM, respectively.

MRM chromatograms of DAPTAD-derivatized 25OHD3-S and 25OHD3-G in human bile. Shown are multiple reaction monitoring chromatograms of DAPTAD-25OHD3-S (A), DAPTAD-d6-25OHD3-S (B), DAPTAD-25OHD3-G (C) and DAPTAD-d6-25OHD3-G (D) in pooled human bile from healthy subjects.
ATP-Dependent Uptake of 25OHD3-G into Inside-Out Plasma Membrane Vesicles.
Detection of 25OHD3-S and 25OHD3-G in human bile suggests that these conjugates are probably formed in the liver and subsequently transported to the bile via efflux transporters on the canalicular membrane of the hepatocytes. To test this possibility, we first performed a rapid screening of hepatic efflux transporters for 25OHD3-S and 25OHD3-G using plasma membrane vesicular transport assays. Major hepatic efflux transporters frequently reported to be involved in the efflux transport of conjugative metabolites were screened using transporter-overexpressing plasma membrane vesicles; these included MRP2 and BCRP on the hepatic canalicular membrane and MRP3 and MRP4 on the sinusoidal membrane. Statistically significant differences in uptake of 50 μM E2-17β-G, a known substrate of these transporters, into plasma membrane vesicles between the ATP and AMP groups were observed with vesicles overexpressing each of the transporters (Fig. 3A). Differences between the ATP and AMP groups associated with the mock control vesicles were also significant but much smaller than those associated with the transporter-overexpressing vesicles. Furthermore, uptake of E2-17β-G into transporter-overexpression vesicles was significantly greater than that into the mock control vesicles in the presence of ATP (Fig. 3A). These data confirmed that the membrane vesicles we used were functional. By incubating 2 μM of 25OHD3-G with membrane vesicles at 37°C for 5 minutes, significant ATP-dependent active uptake of 25OHD3-G by MRP2 and MRP3 was observed (Fig. 3B), suggesting that 25OHD3-G is a substrate of these efflux transporters. On the other hand, there was no significant active uptake of 25OHD3-G by BCRP and MRP4 (Fig. 3B), suggesting that 25OHD3-G is not a substrate of BCRP and MRP4. To further confirm these findings, we performed inhibition studies using selective MRP inhibitors. MK571 (Weiss et al., 2007) and indomethacin (Draper et al., 1997) are relatively selective inhibitors for MRP transporters. We found that the net ATP-dependent uptake of 25OHD3-G at 2 μM into MRP2- and MRP3-expressing plasma membrane vesicles was significantly reduced by 50 μM MK571 to 70% (Fig. 4A) and 50% (Fig. 4B), respectively. Indomethacin at 672 μM also significantly inhibited MRP3-mediated active uptake of 25OHD3-G by ∼75% (Fig. 4B). Results of the inhibition studies provided additional evidence to support the notion that 25OHD3-G can be actively transported by MRP2 and MRP3. Both MK571 and indomethacin also decreased the net active uptake of 25OHD3-G into Sf9 mock membrane vesicles (Fig. 4, A and B), suggesting that Sf9 cells express low levels of endogenous MRP transporters capable of transporting 25OHD3-G.

Screening for ATP-dependent uptake of E2-17β-G and 25OHD3-G in plasma membrane vesicles overexpressing efflux transporters. (A) E2-17β-G, a positive control substrate, was incubated at 50 μM with plasma membrane vesicles overexpressing MRP2, MRP3, BCRP, and MRP4 as well as their corresponding mock control membranes at 37°C for 5 minutes. (B) 25OHD3-G was incubated at 2 μM with plasma membrane vesicles overexpressing MRP2, MRP3, BCRP, and MRP4 as well as their corresponding mock control membranes at 37°C for 5 minutes. Data shown are means ± S.D. of triplicate determinations in a single experiment. Differences between the ATP and AMP groups and between the transporter-overexpression and the mock [Sf9 or human embryonic kidney (HEK)] groups for each transporter were analyzed with Student’s t test; differences with P values of <0.05 were considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001.

ATP-dependent uptake of 25OHD3-G into Sf9 insect cell plasma membrane vesicles overexpressing MRP2 and MRP3. (A) 25OHD3-G at 10 μM was incubated with MRP2-overexpressing Sf9 plasma membrane vesicles or mock control membrane vesicles in the presence or absence of 50 μM MK571 at 37°C for 5 minutes. (B) 25OHD3-G at 3 μM was incubated with MRP3-overexpressing Sf9 plasma membrane vesicles or mock control membrane vesicles in the presence or absence of 50 μM MK571 or 672 μM indomethacin at 37°C for 5 minutes. Differences between the ATP and AMP groups (net ATP-dependent uptake) were calculated and compared between the MK571 or indomethacin and vehicle (dimethyl sulfoxide) treatments for statistical significance by Student’s t test. Data shown are means ± S.D. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, respectively. Representative kinetic profiles of MRP2- and MRP3-mediated ATP-dependent uptake of 25OHD3-G into Sf9 plasma membrane vesicles are shown in (C) and (D), respectively. 25OHD3-G of varying concentrations was incubated with membrane vesicles in the presence of ATP or AMP at 37°C for 5 minutes. The ATP-dependent net uptake into membrane vesicles was plotted against unbound concentrations of 25OHD3-G in the incubations. Data shown are means ± S.D. of triplicate determinations in a single experiment. Unbound Km and Vmax were calculated and presented as means ± S.D. of three independent experiments each with triplicate determinations.
We next estimated kinetic parameters (Km and Vmax) for MRP2- and MRP3-mediated efflux of 25OHD3-G. The representative transport kinetic profiles are shown in Fig. 4C for MRP2, and in Fig. 4D for MRP3. Given high nonspecific binding of 25OHD3-G that can influence kinetic parameter estimation, we first determined unbound fractions of 25OHD3-G in incubation buffers used in the vesicular transport assays, and then used unbound fractions to correct Km values. We found that the unbound fraction Y (%) of 25OHD3-G exhibited a linear relation with the total concentration (X) following the equation: Y = 0.0018X + 7.3119 (r2 = 0.9965) within the concentration range of 0–10 μM, and then reached saturation at ∼25%. After correction with unbound fractions at respective total concentrations, the Km values for MRP2- and MRP3-mediated transport of 25OHD3-G were 1.56 ± 0.43 and 0.25 ± 0.04 μM, and the Vmax were 295.6 ± 87.1 and 66.7 ± 11.6 pmol/mg protein per minute, respectively.
Inhibition of Efflux Transporter Activity by 25OHD3-S.
Owing to high nonspecific binding and passive diffusion, we noticed very high background levels associated with membrane vesicles even after extensive washing with 1.5% BSA or DBP. Such a high background appeared to completely mask activity of efflux transporters for 25OHD3-S with no significant differences in the uptake into membrane vesicles between the ATP and AMP groups (data not shown). Therefore, we were unable to demonstrate whether 25OHD3-S is a substrate of the efflux transporters examined, including BCRP, MRP2, MRP3, and MRP4. To further evaluate the interactions of 25OHD3-S with these transporters, inhibition studies using a model substrate of these transporters (E2-17β-G) were conducted. As shown in Fig. 5, 25OHD3-S effectively inhibited ATP-dependent transport of E2-17β-G by BCRP (Fig. 5A), MRP4 (Fig. 5B), and MRP3 (Fig. 5C) in a concentration-dependent manner, suggesting that 25OHD3-S is a ligand and possibly a substrate of these three transporter proteins. In contrast, 25OHD3-S did not inhibit MRP2-mediated transport of E2-17β-G at all (data not shown). We also determined unbound factions of 25OHD3-S in incubation buffers of the vesicular transport assays and found that the unbound fraction of 25OHD3-S was ∼8% in both HEK293 and Sf9 membrane vesicles and independent of total concentration. After correction for unbound fraction, IC50 values of 25OHD3-S for inhibition of BCRP-, MRP4-, and MRP3-mediated transport of E2-17β-G from duplicate experiments were estimated to be 1.0, 0.96, and 1.88 μM, respectively.

25OHD3-S inhibits ATP-dependent uptake of E2-17β-G into plasma membrane vesicles overexpressing BCRP, MRP4, and MRP3. Representative inhibition profiles of 25OHD3-S for ATP-dependent uptake of E2-17β-G into plasma membrane vesicles overexpressing BCRP (A), MRP4 (B), and MRP3 (C) that were incubated with 25OHD3-S and E2-17β-G at 37°C for 5 minutes. In the incubation, E2-17β-G was at 2 μM for MRP3 and 10 μM for BCRP and MRP4. ATP-dependent net uptake of E2-17β-G (differences between the ATP and AMP groups) was plotted against log10 values of unbound concentrations of 25OHD3-S in membrane vesicle incubations. Data shown are means ± S.D. of triplicate determinations in a single experiment. Unbound IC50 values were estimated from two independent experiments with triplicate determinations in each experiment.
Efflux of 25OHD3-S and 25OHD3-G from Sandwich-Cultured Human Hepatocytes.
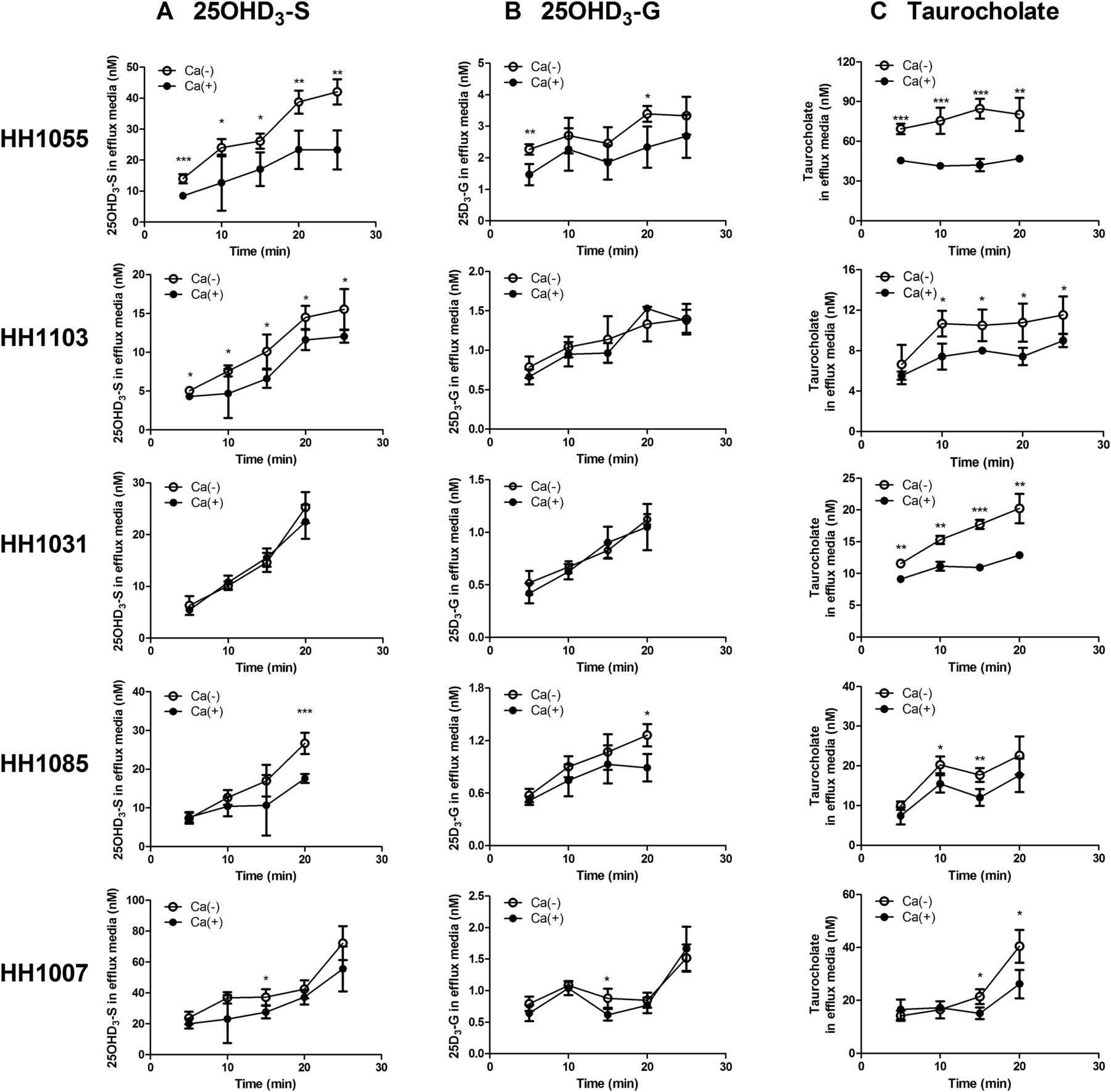
According to results of membrane vesicular transport studies and the detection of 25OHD3 conjugative metabolites in human bile as described above, we hypothesized that 25OHD3-S and 25OHD3-G are transported out of human hepatocytes into the bile after they are converted from 25OHD3 in the liver. To test this hypothesis, we examined biliary efflux of 25OHD3-S and 25OHD3-G using SCHHs. Taurocholate is known to have extensive biliary efflux and therefore was used as a positive control for each preparation of SCHHs. As expected, the efflux of taurocholate from SCHHs in the absence of calcium was significantly higher than in the presence of calcium over time for all five different batches of SCHHs we used with the exception of HH1007 at earlier time points (5 and 10 minutes) (Fig. 6), suggesting that the SCHHs we used were functional.

Efflux of 25OHD3-S, 25OHD3-G, and taurocholate from SCHHs. SCHHs from five different donors (HH1055, HH1103, HH1031, HH1085, and HH1007) were incubated at 37°C with 10 μM 25OHD3 for 4 hours or 2 μM taurocholate for 20 minutes. Efflux of 25OHD3-S (A), 25OHD3-G (B), or taurocholate (C) was then initiated by replacing culture media with Ca2+-containing (sinusoidal efflux, filled circles) or Ca2+-free (sinusoidal + canalicular efflux, open circles) HBSS. Human vitamin D-binding protein (3 μM) was added to the incubation buffer during the efflux phase. Data shown are means ± S.D. for quadruplicate determinations in a single experiment. Differences between with- (filled circles) and without- (open circles) calcium, which represent the canalicular efflux, were analyzed by Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001, respectively.
After incubation of 25OHD3 at 10 μM with SCHHs for 4 hours, we found that 25OHD3-S and 25OHD3-G representing ∼3% of the starting dose of 25OHD3 were formed with a product-formation ratio of around 30:1 (25OHD3-S/25OHD3-G), calculated on the basis of their recovery from efflux incubation buffers and cell lysates of SCHH. Both 25OHD3-S and 25OHD3-G rapidly appeared in the efflux buffer in the absence and presence of calcium and their concentrations increased with time, suggesting a time-dependent passive or active efflux of the conjugates from hepatocytes (Fig. 6). We noticed substantial interindividual variations in both sinusoidal and canalicular efflux of 25OHD3-S and 25OHD3-G among SCHHs from five different donors. The increases in concentrations of 25OHD3-S and 25OHD3-G in efflux buffers over time (0–20 minutes) in the presence of calcium, which represent sinusoidal efflux rates, followed the rank orders of HH1007 > HH1055 > HH1031 > HH1085 > HH1103 and of HH1055 > HH1007 > HH1103 > HH1031 > HH1085 for 25OHD3-S and 25OHD3-G, respectively. In the absence of calcium, concentrations of 25OHD3-S in efflux buffers over time were significantly higher than those in the presence of calcium in SCHHs from donors HH1055, HH1103, HH1085, and HH1007, but not HH1031. In particular, SCHHs from the donor HH1055 showed significantly higher canalicular efflux activity of 25OHD3-S than other donors. Likewise, concentrations of 25OHD3-G in efflux buffers over time in the absence of calcium were significantly higher than those in the presence of calcium in SCHHs from donors HH1055, HH1085 and HH1007, but not HH1103 and HH1031. The donor HH1031 did not exhibit canalicular efflux of either 25OHD3-S nor 25OHD3-G, despite high canalicular efflux of taurocholate.
To confirm if BCRP or MRPs are involved in the canalicular and sinusoidal efflux of 25OHD3-S and 25OHD3-G, FTC, a selective BCRP inhibitor, or MK571, a selective MRP inhibitor, was added to incubation buffer during the loading and efflux phases. SCHHs from the donor HH1055, which showed the highest efflux activity among all the donors analyzed, were used in the inhibition studies. As expected, the canalicular efflux of 25OHD3-S was significantly decreased by the addition to FTC compared with control with no FTC added. We calculated biliary efflux index in the presence and absence of FTC. The mean BEI (%) of 25OHD3-S from three independent experiments was significantly decreased by FTC at 15 and 20 minutes of efflux (Table 1), confirming that the biliary efflux of 25OHD3-S is probably mediated by BCRP. However, the effects of MK571 on the efflux of 25OHD3-G and 25OHD3-S cannot be analyzed. Owing to the near complete inhibition on both the glucuronidation and sulfation of 25OHD3 in the hepatocytes by MK571 (data not shown), the two conjugative metabolites were undetectable in most of the efflux buffers analyzed.
Biliary excretion index of 25OHD3-S in SCHHs in the presence and absence of the BCRP inhibitor FTC
SCHHs were incubated at 37°C with 25OHD3 (10 μM) for 4 hours in the presence or absence of FTC (10 μM), and the efflux was initiated by replacing the culture media with Ca2+-containing (sinusoidal efflux) or Ca2+-free (sinusoidal + canalicular efflux) HBSS in the presence or absence of FTC (10 μM). Human vitamin D-binding protein (3 μM) was added during the efflux phase. Data shown are means ± S.D. of three independent experiments, and each experiment was performed in quadruplicate. Differences between the control and FTC-inhibited groups were analyzed by Student’s t test.
Cellular Uptake of 25OHD3-G and 25OHD3-S by OATPs.
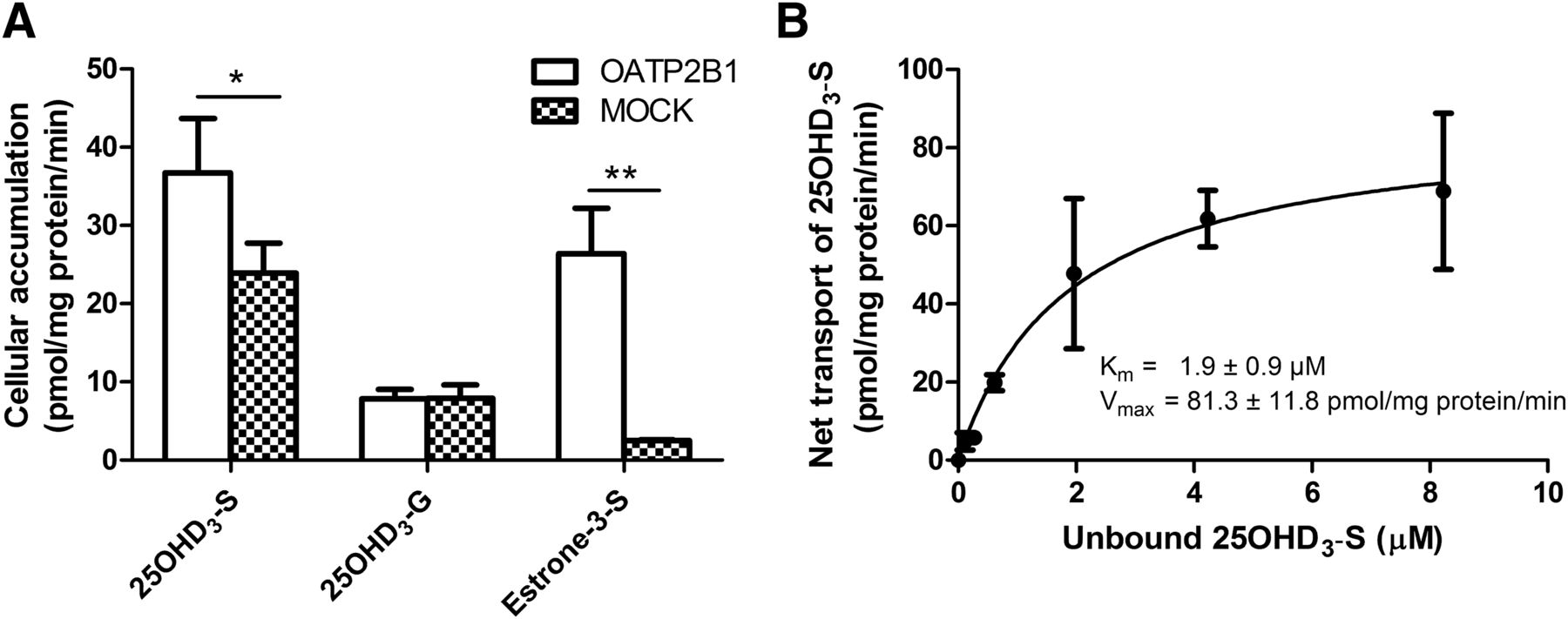
OATP2B1 is a major uptake transporter in enterocytes and facilitates in intestinal absorption of xenobiotics (Tamai, 2012). Once in the bile, 25OHD3-G and 25OHD3-S could be excreted to the intestinal lumen and be absorbed into the enterocytes by uptake transporters. In addition, 25OHD3-G and 25OHD3-S in the circulation may be transported back into the hepatocytes by uptake transporters on the sinusoidal membrane, such as OATP1B1 and OATP1B3. We therefore performed cellular uptake studies to show if 25OHD3-G and 25OHD3-S are substrates of OATP2B1, OATP1B1, and OATP1B3. E1-3-S was used as a positive control, as it is a known substrate of OATP2B1. As expected, cellular accumulation of E1-3-S in Flp-In-CHO/OATP2B1 cells was approximately 10 times greater than that in mock CHO control cells (Fig. 7A). We found that cellular accumulation of 25OHD3-S in Flp-In-CHO/OATP2B1 cells was significantly increased by ∼50% compared with that in mock CHO cells (Fig. 7A); however, there were no significant differences in uptake of 25OHD3-G into OATP2B1 cells and the mock control cells (Fig. 7A). The results indicate that 25OHD3-S, but not 25OHD3-G, is a substrate of OATP2B1. Further kinetic measurements revealed an unbound Km of 1.9 ± 0.9 μM and a Vmax of 81.3 ± 11.8 pmol/mg protein per minute for OATP2B1-mediated uptake of 25OHD3-S from five independent experiments. A representative kinetic profile of OATP2B1-mediated uptake of 25OHD3-S is shown in Fig. 7B.

Uptake of 25OHD3-S and 25OHD3-G by OATP2B1. (A) 10 μM of 25OHD3-S or 25OHD3-G or 10 μM of E1-3-S was incubated with OATP2B1-overexpressing or mock control CHO cells at 37°C for 5 minutes. Differences in cellular accumulation of 25OHD3-S, 25OHD3-G or E1-3-S between OATP2B1-transfected and nontransfected cells were analyzed by Student’s t test. *P < 0.05; **P < 0.01, respectively. Data shown are means ± S.D. of three determinations in a single experiment. Consistent results were obtained in another two independent experiments. (B) A representative kinetic profile of OATP2B1-mediated uptake of 25OHD3-S. Net uptake was calculated by subtracting cellular accumulation of 25OHD3-S in mock control cells from that in OATP2B1-overexpressing cells and plotted against unbound concentrations of 25OHD3-S in incubation buffers. Data shown are means ± S.D. of triplicate determinations in a single experiment. Km and Vmax were estimated from five independent experiments with triplicate determinations in each experiment.
Furthermore, cellular accumulation of 25OHD3-S in OATP1B3-overexpressing CHO cells was about 2-fold (P < 0.001) that in wild-type CHO parent cells and this difference was completely abolished by the addition of 20 μM CsA, a potent OATP inhibitor (Fig. 8A). On the other hand, there were no differences in cellular accumulation of 25OHD3-S in OATP1B1-overexpressing cells and the wild-type CHO parent cells (Fig. 8A). The data indicate that 25OHD3-S is a substrate of OATP1B3, but not OATP1B1. Likewise, cellular accumulation of 25OHD3-G in OATP1B1- and OATP1B3-overexpressing cells was approximately 4-fold (P < 0.001) and 25-fold (P < 0.001) greater than that in the wild-type CHO parent cells, respectively, and this difference was completely diminished by CsA (Fig. 8B). Therefore, 25OHD3-G is a substrate of both OATP1B1 and OATP1B3.

Uptake of 25OHD3-S and 25OHD3-G by OATP1B1 and OATP1B3. 25OHD3-S (A) or 25OHD3-G (B) at 2 μM was incubated with CHO/OATP1B1, CHO/OATP1B3, or wild-type (WT) parent control CHO cells at 37°C for 5 minutes in the presence or absence of CsA at 20 μM. Differences in cellular accumulation of 25OHD3-S (A) or 25OHD3-G (B) between OATP-overexpressing cells and wild-type parent control cells were analyzed by Student’s t test. **P < 0.01; ***P < 0.001, respectively. Data shown are means ± S.D. of four determinations in a single experiment. Consistent results were obtained in another two independent experiments.
Discussion
The present study demonstrates active transport of 25OHD3-S and/or 25OHD3-G by MRP2, MRP3, MRP4, BCRP, OATP1B1, OATP1B3, or OATP2B1. Owing to the lipophilic side chain at carbon C20, 25OHD3-S and 25OHD3-G were highly bound to cell membrane and plastic surface. This resulted in high background levels in membrane vesicular and cellular accumulation assays that masked real active transport, and initially no direct uptake could be observed for either 25OHD3-S or 25OHD3-G on our first try. To reduce nonspecific binding, we optimized washing steps with a final washing buffer containing 1.5% BSA (w/v) that were repeated five and two times for membrane vesicle and intact cell, respectively. Addition of DBP offered no further advantage (data not shown). These conditions allowed us to quantify direct active uptake of 25OHD3-G into membrane vesicles and transfected cells, and active uptake of 25OHD3-S into transfected cells, but not membrane vesicles. Although we did not observe ATP-dependent transport of 25OHD3-S into membrane vesicles, we found that 25OHD3-S was a potent inhibitor of E2-17β-G transport by BCRP, MRP3, and MRP4, indicating that 25OHD3-S is a ligand and possibly a substrate of these transporters.
Dehydroepiandrosterone sulfate (DHEAS) is a steroid sulfate conjugate with a structure similar to 25OHD3-S, except for the lipophilic side chain at carbon C20. DHEAS has been shown to be a potent inhibitor of MRP1, MRP4, and BCRP, and a substrate of these transporters by direct vesicular uptake (Suzuki et al., 2003; Zelcer et al., 2003). Considering the similar structures of 25OHD3-S and DHEAS, it is plausible that they share comparable transporter-binding properties. Therefore, inhibition of transporter activities by 25OHD3-S, which was observed in the current study, is most probably competitive in nature, and 25OHD3-S is possibly a substrate of MRP3, MRP4, and BCRP. Whether active transport of 25OHD3-S is of physiologic importance depends on the extent of passive diffusion versus nonspecific sequestration in the membrane vesicle systems, something we have not been able to address at this time. The failure to show direct uptake of 25OHD3-S into membrane vesicles could have been the result of high membrane permeability driven by high lipophilicity [LogP: 7.386, calculated using ACD/Labs Software V11.02 (Toronto, ON, Canada)] compared with DHEAS (LogP: 3.522), much smaller inner space volume of membrane vesicles compared with intact cells, as well as high levels of protein binding. These resulted in high background levels associated with membrane vesicles that could not be reduced by extensive wash with high concentrations of BSA and DBP.
To confirm biliary transport in a more physiologic model, efflux of 25OHD3-S and 25OHD3-G were investigated in SCHHs. There are theoretically two ways to load the compounds in this system: 1) directly load 25OHD3-S and 25OHD3-G and determine the efflux after removing excessive free compounds and 2) incubate 25OHD3 with SCHHs to generate 25OHD3-S and 25OHD3-G, and then observe the efflux of produced metabolites. Direct loading appears to be straightforward, time-saving, and not affected by metabolite formation activity of the hepatocyte. However, after several attempts, we found that excessive unbound conjugates from the loading step could not be sufficiently removed, even after extensive washing with BSA-containing media, to permit quantitation of net active transport. Therefore, the second method was adopted, and the conjugates were generated de novo from 25OHD3 in SCHHs before assessment of hepatic efflux. Of course, this had the advantage of better mimicking in vivo hepatic disposition of the 25OHD3 conjugates. We also found that it was necessary to include DPB in the efflux buffer to facilitate release of 25OHD3-S and 25OHD3-G from cell surfaces during the efflux phase, something that is also expected in vivo for basolateral transport. Without DBP, only minimal amount of 25OHD3-S and 25OHD3-G could be detected in the efflux buffer, which is consistent with the competing affinity of 25OHD3-S and 25OHD3-G for cell membranes or plastic surface. However, it is difficult to predict what happens during biliary transport on the basis of our in vitro observation. Bile does not contain DBP, but it is rich in bile salts, which are good surfactants and might greatly enhance the solubility and affinity of lipophilic 25OHD3-S and 25OHD3-G conjugates in bile.
We observed substantial interindividual variations of both sinusoidal and canalicular efflux of 25OHD3-S and 25OHD3-G in SCHHs from five different donors, which could have been caused by interindividual variations in efflux transporter expression. However, owing to the limitation of method sensitivity and sample amount available, quantification of protein levels of efflux transporters in the SCHHs by LC-MS-based proteomics could not be achieved (data not shown). To evaluate the roles of MRPs and BCRP in the efflux of 25OHD3-S and 25OHD3-G in SCHHs, relatively specific inhibitors were added to chemically knock out the corresponding transporter. In our SCHH efflux assays, 25OHD3-S and 25OHD3-G were formed in hepatocytes during the 4-hour loading phase with 25OHD3, during which the conjugates could be effluxed into bile canaliculi immediately upon formation. Therefore, to completely block the efflux, the transporter inhibitor was added at the beginning of both loading and efflux phases. The BCRP-specific inhibitor FTC significantly reduced the canalicular efflux of 25OHD3-S, suggesting an essential role of BCRP in the biliary efflux of 25OHD3-S from the liver. To inhibit MRPs, we used MK571, the most commonly used MRP inhibitor. However, we found that MK571 had a profound effect on the conjugative enzymes, essentially abolishing 25OHD3-S and 25OHD3-G formation from 25OHD3. This has also been observed by other researchers (Barrington et al., 2015). Owing to low metabolic formation, the roles of MRPs in the efflux of 25OHD3-S and 25OHD3-G in SCHHs could not be evaluated. We recognize that the concentration of 25OHD3 (10 μM) we used in experiments with SCHHs is not a physiologic exposure. Lower concentrations could not be used because of analytical limitations. However, given the very long half-life of 25OHD3 in humans (14 days), we would suggest that hepatic 25OHD3 conjugation and biliary secretion in vivo is a slow but physiologically important process, even at low (50 nM) physiologic concentrations of 25OHD3.
The absence of 25OHD3-S and 25OHD3-G in urine, despite significant circulating concentrations (Wong et al., 2018), suggests that there is an effective means for hepatic reuptake of 25OHD3-S and 25OHD3-G. OATP1B1, OATP2B1, and OATP1B3 are the major uptake transporters on the sinusoidal membrane of hepatocytes (Badée et al., 2015). Besides the liver, OATP2B1 is also expressed in many other tissues, including intestinal epithelial cells (Gröer et al., 2013). We showed that 25OHD3-S is a good substrate of OATP2B1 and OATP1B3, providing a mechanism for excretion from the body through the liver. This also offers a mechanism for intestinal absorption of 25OHD3-S, followed by excretion into the bile (by BCRP). Similarly, the excretion of 25OHD3-G from the body may be initiated by OATP1B1/OATP1B3-mediated hepatic uptake, followed by MRP2-mediated biliary excretion. Although 25OHD3-G was not transported by OATP2B1, it might be deconjugated by intestinal bacteria, which are rich in glucuronidase and can hydrolyze diverse types of glucuronide conjugates (Kim and Jin, 2001; Gao et al., 2011). Generated 25OHD3 could then be reabsorbed from the intestinal lumen by passive diffusion.
Considering that 25OHD3-S (Wong et al., 2018) and 25OHD3-G (Wang et al., 2014) are strongly bound to DBP and that DBP is produced in the liver, it is possible that the conjugates formed in the liver could also be effluxed into blood out of the liver bound to the DBP, following protein synthesis in the liver. This mechanism was postulated for other lipophilic vitamins, such as vitamin E (Drevon, 1991) and vitamin K (Shearer et al., 2012), which are transported out of the liver bound to lipoproteins. Whether this occurs for 25OHD3 conjugates is unknown and remains to be investigated.
In summary, as shown in Fig. 9, the present study revealed that 25OHD3-G is a substrate of MRP2, MRP3, OATP1B1, and OATP1B3, and that 25OHD3-S is a substrate of OATP2B1 and OATP1B3, and possibly a substrate of BCRP, MRP3, and MRP4. Thus, in the liver, 25OHD3-G and 25OHD3-S could be excreted into the bile though MRP2 and BCRP, respectively, and back into the blood circulation by MRP3 (both) and MRP4 (25OHD3-S). The sinusoidal OATP transporters may pump unbound 25OHD3-G and 25OHD3-S from the blood into hepatocytes for further biliary excretion or blood recirculation. In the intestinal tract, OATP2B1 may mediate transport of 25OHD3-S into proximal enterocytes, following biliary excretion. Parts of the model presented in Fig. 9 related to the intestinal disposition and downstream effects of 25OHD3-G and 25OHD3-S are speculative and not yet supported by experimental evidence. We present it to stimulate further research and hypothesis testing. For example, can 25OHD3-S and 25OHD3-G be deconjugated to 25OHD3 by intestinal sulfatases or glucuronidases? Would this occur in the lumen of the gastrointestinal traction or after transporter-mediated uptake into enterocytes? What is the kinetics of 25OHD3 metabolism to 1α,25-(OH)2D3 by CYP27B1 in the intestinal tract and would in vivo conversion rates enhance enterocyte levels of 1α,25-(OH)2D3 following biliary secretion of 25OHD3 conjugates? Lastly, the relative importance of the conjugative pathways versus the oxidation pathways (shown in Fig. 1) in the systemic clearance of 25OHD3 remains unclear, as is the contribution of enterohepatic circulation of the 25OHD3 conjugates to the overall disposition and biologic activity of vitamin D. These are important topics of future investigation and the present study represents the first step in addressing this knowledge gap.

A proposed model of enterohepatic transport of 25OHD3-S and 25OHD3-G in the liver and intestine. Solid lines represent confirmed routes; dashed lines represent routes to be confirmed in future studies.
Acknowledgments
We would like to thank Dr. Danny D. Shen and Brian R. Phillips in the Department of Pharmaceutics, University of Washington, Seattle, WA for sharing LC-MS instruments and assistance in the analytical method development. We also greatly thank Drs. Bruno Stieger and Bruno Hagenbuch for their generous gifts of cell lines.
Authorship Contributions
Participated in research design: Gao, Thummel, Mao.
Conducted experiments: Gao, Liao, Han.
Performed data analysis: Gao.
Wrote or contributed to the writing of the manuscript: Gao, Liao, Han, Thummel, Mao.
Footnotes
- Received October 9, 2017.
- Accepted February 15, 2018.
This work was supported by the National Institutes of Health [Grant GM063666].
Abbreviations
- 1α,25-(OH)2D3
- 1α,25-dihydroxyvitamin D3
- 25OHD3-G
- 25OHD3-3-O-glucuronide
- 25OHD3-S
- 25OHD3-3-O-sulfate
- 25OHD3
- 25-hydroxyvitamin D3
- BCRP
- breast cancer resistance protein
- BEI
- biliary efflux index
- BSA
- bovine serum albumin
- CHO
- Chinese hamster ovary
- CsA
- cyclosporine A
- DAPTAD
- 4-(4′-dimethylaminophenyl)-1,2,4-triazoline-3,5-dione
- DBP
- vitamin D-binding protein
- DHEAS
- dehydroepiandrosterone sulfate
- E1-3-S
- estrone-3-sulfate
- E2-17β-G
- estradiol-17β-glucuronide
- E2-3-S
- estradiol-3-sulfate
- FTC
- the BCRP inhibitor fumitremorgin C
- HBSS
- Hanks’ balanced salt solution
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MRP
- multidrug resistance protein
- OATP
- organic anion transporting polypeptide
- P450
- cytochrome P450
- SCHH
- sandwich-cultured human hepatocyte
- VDR
- vitamin D receptor
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}