


Abstract
To estimate the clinical impact of pharmacokinetic modulation via breast cancer resistance protein (BCRP), in vivo approaches in nonclinical settings are desired in drug development. Clinical observation has identified curcumin as a promising candidate for in vivo selective BCRP inhibition, in addition to several well known inhibitors, such as lapatinib and pantoprazole. This study aimed to confirm the inhibitory efficacy of curcumin on gastrointestinal BCRP function in cynomolgus monkeys and to perform comparisons with lapatinib and pantoprazole. Oral area under the plasma concentration-time curve (AUC) and bioavailability of well known BCRP (sulfasalazine and rosuvastatin), P-glycoprotein (fexofenadine, aliskiren, and talinolol), and CYP3A (midazolam) substrates were investigated in the presence and absence of inhibitors. Oral exposures of sulfasalazine and rosuvastatin were markedly elevated by curcumin with minimal changes in systemic clearance, whereas pharmacokinetic alterations after fexofenadine, aliskiren, and talinolol oral exposure were limited. Curcumin increased oral midazolam exposure without affecting systemic clearance, presumably owing to partial inhibition of intestinal CYP3A. Lapatinib increased the oral AUC for sulfasalazine to a greater extent than curcumin did, whereas pantoprazole had a smaller effect. However, lapatinib also exerted significant effects on fexofenadine, failed to selectively discriminate between BCRP and P-glycoprotein inhibition, and had an effect on oral midazolam exposure comparable with that of curcumin. Thus, pharmacokinetic evaluation in monkeys demonstrated that pretreatment with curcumin as an in vivo selective BCRP inhibitor was more appropriate than pretreatment with lapatinib and pantoprazole for the assessment of the impact of BCRP on gastrointestinal absorption in nonrodent models.
Introduction
Breast cancer resistance protein (BCRP, ABCG2) is an important ATP-binding cassette efflux transporter related to drug pharmacokinetics (PK) and uric acid homeostasis; some ABCG2 polymorphisms decrease membrane transport protein expression levels (Woodward et al., 2009; Mao and Unadkat, 2015). Indeed, the ABCG2 421C>A variant is the best known; the oral area under the plasma concentration-time curve (AUC) of BCRP substrates [sulfasalazine (SASP): 2.1–3.5-fold (Urquhart et al., 2008; Yamasaki et al., 2008; Adkison et al., 2010; Gotanda et al., 2015); rosuvastatin (RSV): 1.6–3.2-fold (Zhang et al., 2006; Keskitalo et al., 2009b; Zhou et al., 2013; Birmingham et al., 2015; Wan et al., 2015); fluvastatin: 1.7-fold (Keskitalo et al., 2009a); atorvastatin: 1.3–1.7-fold (Keskitalo et al., 2009b; Birmingham et al., 2015); and sunitinib: 2.5-fold (Mizuno et al., 2012)] increased in ABCG2 421AA subjects compared with that in ABCG2 421CC subjects, which led to a higher incidence of adverse events and overefficacy of RSV (Lee et al., 2013), fluvastatin (Miroševic Skvrce et al., 2013), atorvastatin (Mirošević Skvrce et al., 2015), and sunitinib (Miura et al., 2014; Low et al., 2016).
It is therefore desirable to establish in vivo evaluation in nonclinical settings that enable the simple estimation of the clinical impact of BCRP on drug disposition, which is much more pronounced for orally than intravenously administered drugs, because some BCRP substrates [e.g., nitrofurantoin (Merino et al., 2005) and pitavastatin (Ieiri et al., 2007)] show limited PK changes regardless of ABCG2 variants. Previously, we demonstrated that cynomolgus monkeys pretreated with elacridar (EL) as a BCRP inhibitor and Abcg2-deficient mice were useful for in vivo evaluation of the clinical impact of BCRP on drug absorption (Karibe et al., 2015). However, further discovery studies for BCRP inhibitors more selective than EL are required for the adequate assessment of the intestinal BCRP contribution in monkeys, as EL is also a strong P-glycoprotein (P-gp) inhibitor (Matsson et al., 2009).
As indicated by the US Food and Drug Administration, (http://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm), curcumin is a BCRP inhibitor, like cyclosporine A and eltrombopag, which increased human RSV exposure presumably through inhibition of both BCRP and organic anion-transporting polypeptide (OATP). Kusuhara et al. (2012) reported that the oral AUC increase of SASP caused by curcumin pretreatment in ABCG2 421CC subjects (3.2-fold) was comparable to that observed clinically in ABCG2 421C>A subjects (2.1–3.5-fold). This clinical observation could be inferred from curcumin increasing the oral exposure of SASP in wild-type mice, whereas Abcg2-deficient mice were unaffected (Shukla et al., 2009; Kusuhara et al., 2012). Aside from curcumin, Lee et al. (2015) proposed the potential usefulness of lapatinib (LAP) at one-fifth the approved dosage (250 mg) for in vitro assessment of clinical BCRP-mediated drug-drug interactions (DDIs). Pantoprazole (PAN) inhibited BCRP in vitro (Elsby et al., 2016) and modulated sunitinib PK in rats at 40 mg/kg (Kunimatsu et al., 2013), although the human PK of SASP and RSV were not altered in any genotypic cohort by coadministration of 40 mg PAN (Adkison et al., 2010; Huguet et al., 2016), possibly owing to the large difference in dosage administered to humans and rats.
However, concerns remain about in vivo inhibition of transporters and metabolic enzymes other than BCRP, as shown by EL, cyclosporine A, and eltrombopag. Curcumin inhibited P-gp, multidrug resistance-associated protein 2, OATP1B1, OATP1B3, OATP2B1, CYP3A, and phase II enzymes in vitro and in vivo (Appiah-Opong et al., 2007; Juan et al., 2007; Volak et al., 2008; Lee et al., 2011; He et al., 2012; Kusuhara et al., 2012; Ge et al., 2016; Sun et al., 2016; Zhou et al., 2017). LAP increases human AUCs of P-gp (digoxin), CYP3A4 [midazolam (MDZ)], and CYP2C8 substrates (paclitaxel) at its approved dosage (https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/022059s022lbl.pdf). In contrast, there are few reports on the clinical DDI risk of PAN (https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020987s050,022020s012lbl.pdf).
This study aimed to confirm, in a preclinical model using cynomolgus monkeys, the appropriateness of curcumin as an in vivo BCRP inhibitor, with respect to inhibition of gastrointestinal BCRP function as well as intestinal and hepatic OATPs, P-gp, and CYP3A activity. Model substances described in the US Food and Drug Administration Draft Guidance [Center for Drug Evaluation and Research (CDER), 2012], including probes of BCRP (SASP and RSV), P-gp [fexofenadine (FEX), talinolol (TLN), and aliskiren (AL)], and CYP3A (MDZ), were used. Comparative studies with LAP and PAN were also performed. To elucidate the cause of observed AUC changes, the DDI potentials of BCRP inhibitors were assessed by in vitro studies.
Materials and Methods
Materials
RSV calcium, TLN, 1′-hydroxymidazolam, 4′-hydroxymidazolam, curcumin β-d-glucuronide (CG), and curcumin sulfate (CS) tetrabutylammonium salt were purchased from Toronto Research Chemicals Inc. (North York, Canada). AL for in vitro studies was obtained from AdooQ BioScience (Irvine, CA). Novobiocin sodium, rifampicin, and SASP were purchased from Sigma-Aldrich (St. Louis, MO). AL hemifumarate for in vivo studies, FEX hydrochloride, ketoconazole (KTZ), and PAN sodium were purchased from LKT Laboratories, Inc. (St. Paul, MN). Curcumin and MDZ were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Newly developed nanoparticulate curcumin with increased water solubility (Theracurmin) was purchased from Theravalues Corporation (Tokyo, Japan). LAP was purchased from LC Laboratories (Woburn, MA). Cryopreserved cynomolgus monkey hepatocytes were obtained from BioreclamationIVT (Baltimore, MD). Pooled liver and small intestinal microsomes from humans and male cynomolgus monkeys were purchased from Xenotech, LLC (Lenexa, KS). [3H]RSV calcium (10 Ci/mmol) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO). [3H]Estrone sulfate ammonium salt (ES; 1 Ci/mmol) and [3H]digoxin (1 Ci/mmol) were obtained from PerkinElmer, Inc. (Waltham, MA). All other chemicals and reagents were of analytical grade and available from commercial sources.
In Vivo Studies
Animals.
The in vivo studies, with a minimum drug washout period of at least 2 weeks, were performed by Narita Animal Science (Chiba, Japan). Male cynomolgus monkeys (aged 3–8 years) were supplied by Guangxi Grandforest Scientific Primate Co., Ltd (Guangxi, China). All animals were housed in a temperature- and humidity-controlled room with a 12-hour light cycle, fed a standard animal diet as appropriate for each species, and given ad libitum access to water. All experimental procedures were conducted in accordance with the in-house guidelines of the Institutional Animal Care and Use Committee of Narita Animal Science and Daiichi Sankyo Co., Ltd.
Pharmacokinetic Studies in Cynomolgus Monkeys with or without Pretreatment of Curcumin, Lapatinib, or Pantoprazole.
Cynomolgus monkeys (body weight 2.6–5.9 kg) were fasted overnight, but given free access to water prior to drug administration. First, curcumin in the form of Theracurmin, a nanoparticulate colloidal dispersion with improved oral bioavailability and high water solubility and stability in UV light and heat (Sasaki et al., 2011), was administered by oral gavage at 30 mg/kg (or 0 mg/kg in the case of the placebo) as a well triturated suspension in 0.5% aqueous methylcellulose (MC). The dose was set on the basis of a previous human BCRP DDI study (Kusuhara et al., 2012). Ten minutes after pretreatment, the BCRP substrates (SASP and RSV), P-gp (FEX, TLN, and AL), and CYP3A (MDZ) were administered by oral gavage at 5, 1, 2, 1, 3, and 2 mg/kg, respectively; additionally, SASP, RSV, and MDZ were intravenously (IV) administered at 5, 1, and 0.2 mg/kg, respectively. SASP, FEX, TLN, AL, and MDZ for oral administration (PO) were prepared as suspensions in 0.5% MC, and SASP and MDZ for IV administration were prepared as solutions in 1% sodium hydrogen carbonate and in saline modified to a weak acidic pH by 0.1 M hydrogen chloride, respectively. The dosing solution of RSV was prepared with 5% dimethylacetamide in saline. Meanwhile, 10 minutes before the administration of P-gp substrates and MDZ, EL (5 mg/kg) and KTZ (4 mg/kg) in 0.5% MC were administered by oral gavage instead of curcumin as positive controls for the inhibition of intestinal P-gp and CYP3A, respectively, in cynomolgus monkeys (Ward and Azzarano, 2004; Ogasawara et al., 2007).
The human PK properties of the probes selected in this study are summarized in Table 1. SASP, RSV, FEX, TLN, and AL were hardly metabolized in vivo. MDZ, which is widely used as a CYP3A probe in clinical DDI studies, was not affected by inhibition of BCRP, P-gp, or hepatic OATPs (CDER, 2012; Maeda, 2015). Moreover, all substrates evaluated in this study (except for TLN) were confirmed to have in vivo intestinal DDI potential via BCRP, P-gp, and CYP3A in monkey PK studies, and showed similar DDI effects in humans (Ogasawara et al., 2007; Karibe et al., 2015; Tsukimoto et al., 2015).
Human pharmacokinetic properties, metabolizing enzymes, and transporters of probes tested in this study
The experimental conditions for pretreatment with LAP or PAN were the same as those for curcumin. Briefly, 10 minutes after PO administration of placebo or LAP (5 mg/kg) as a well triturated suspension in 0.5% MC, SASP, FEX, and MDZ were administered by oral gavage, or SASP and MDZ were intravenously administered. The dosage of oral pretreatment with LAP (5 mg/kg) was determined from the recommended human dose in a BCRP DDI study by Lee et al. (2015). PAN was administered by oral gavage as a solution at 0.6 or 20 mg/kg in 0.5% MC for 10 minutes prior to intravenous or oral administration of SASP. The PAN doses were set at 0.6 and 20 mg/kg according to reported clinical [no significant human AUC change (Adkison et al., 2010; Huguet et al., 2016)] and nonclinical [significant rat AUC increase (Kunimatsu et al., 2013)] doses, respectively.
Blood samples were collected upon pretreatment, as well as 0.0833 (for IV-administered group only), 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after dosing of each probe substrate. The plasma samples were separated by centrifugation (4°C, 21,900g, 3 minutes, anticoagulant: heparin sodium) and stored at −20°C until analysis. Each study was performed using the same monkeys for each administration route with a 2-week washout period. For curcumin, each group comprised 4–10 animals. For LAP and PAN, the same three and four monkeys were used for IV and PO, respectively.
In Vitro Studies
Efflux Transporter Inhibition Assays in Caco-2 Cells.
The inhibition potentials of BCRP and P-gp by curcumin, LAP, and PAN were evaluated with Caco-2 cells obtained from the American Type Culture Collection (ATCC accession number CRL-2102; Manassas, VA). In addition to curcumin, the main curcumin metabolites (CG and CS) were also assessed, because plasma concentrations of curcumin are almost undetectable after oral administration of curcumin to humans, whereas its metabolites are found in peripheral and portal circulation (Garcea et al., 2004; Vareed et al., 2008). The experimental conditions were selected as previously described (Karibe et al., 2015). In brief, Caco-2 cells were grown on HTS Transwell 24-well plates (0.4 μm polycarbonate membrane; culture surface area 0.33 cm2; Corning Inc., Corning, NY) at 5 × 104 cells per well for 15–17 days, with culture medium exchange every 3–4 days. All monolayers used in the assay exhibited transepithelial electrical resistance values greater than 300 Ω × cm2.
Before the assay, the culture medium on both the apical and basal sides was exchanged for assay buffer (Hanks’ balanced salt solution supplemented with 10 mM HEPES adjusted to pH 7.4), and the cells were washed by preincubation for at least 20 minutes at 37°C. The preincubated buffer on the apical or basal side (the donor side) was replaced with or without test compounds [final dimethyl sulfoxide (DMSO) concentration: 1%] containing each of the radiolabeled drugs ([3H]RSV, [3H]ES, and [3H]digoxin) after the buffer on the opposite side (the receiver side) was replenished with assay buffer. Simultaneously, to assure BCRP and P-gp inhibition in the assay, the vectorial transport of the test compounds was evaluated in the presence of 100 μM novobiocin and verapamil, respectively. After a 120-minute incubation at 37°C, aliquots of the solutions were sampled from the receiver side, mixed with scintillation cocktails, and their radioactivities determined using a liquid scintillation counter. The counting was corrected by an external standard source method, and the radioactivity in the samples was calculated by subtracting background radioactivity from measured radioactivity in the samples.
To evaluate the inhibition of SASP, FEX, TLN, AL, or MDZ efflux, assay buffer containing 5 μM DMSO solutions was used instead of radiolabeled test compounds (final DMSO concentration, 0.05%). After incubation for 120 minutes at 37°C, aliquots of the solutions were also sampled from the receiver side and stored at −20°C until quantification by liquid chromatography–tandem mass spectrometry (LC-MS/MS).
The incubations for apical-to-basal and basal-to-apical transport were performed in triplicate. The amount of each substrate transported across the monolayer was calculated on the basis of the value of the transported concentrations of the substrates multiplied by the volume, and the apparent permeability coefficient (Papp) was calculated from eq. (1): (1)where dQ/dt, A, and C0 represent the amounts of the test substrates transported within a given time period, the surface area of the monolayer, and the initial concentrations of the substrates, respectively. After the mean
(1)where dQ/dt, A, and C0 represent the amounts of the test substrates transported within a given time period, the surface area of the monolayer, and the initial concentrations of the substrates, respectively. After the mean  values from triplicate data were determined, the efflux ratio (ER) and remaining activity (% of control) (RA) were calculated using eqs. (2) and (3), respectively:
values from triplicate data were determined, the efflux ratio (ER) and remaining activity (% of control) (RA) were calculated using eqs. (2) and (3), respectively:
 (2)
(2) (3)
(3)CYP3A Inhibition Assays Using Liver and Small Intestinal Microsomes from Cynomolgus Monkeys and Humans.
The assessment of curcumin, CG, and CS as potential inhibitors of CYP3A, the major cytochrome P450 enzyme in the small intestine of monkeys, was performed using liver and small intestinal microsomes from cynomolgus monkeys and humans by monitoring the 1′-hydroxylation and 4′-hydroxylation of midazolam. Briefly, pooled microsomes were reconstituted at 0.1 mg/ml protein in 100 mM phosphate buffer (pH 7.4) and preincubated with 4 μM MDZ and inhibitors (0.5–50 μM) with DMSO at a final concentration of 0.1% at 37°C for 5 minutes. The assays were then initiated by the addition of an NADPH-generating system (25 mM G6P, 0.5 IU/ml G6PDH, 10 mM MgCl2, 2.5 mM NADP+). Meanwhile, KTZ (final concentration, approximately 1 μM), a potent CYP3A inhibitor, was tested to confirm CYP3A inhibition in the assay. After incubation for 10 minutes at 37°C, an aliquot of the reaction mixture was placed in a centrifuge tube containing ice-cold methanol and acetonitrile with an internal standard to stop the reaction. The incubations were performed in duplicate. When CYP3A inhibition potential was observed (%inhibition compared with no inhibitor: below 50%), estimations of the inhibition constant (Ki) were performed using the Km,app method [Kakkar et al., 1999: eqs. (4), (5), and (6)] in the same microsomal reaction with several MDZ concentrations (1–16 μM) and inhibitors (1–100 μM) using Phoenix WinNonlin (ver. 6.3; Pharsight Corporation, Mountain View, CA). All samples were analyzed by LC-MS/MS. (4)
(4) (5)
(5) (6)where S is the MDZ concentration, I is the inhibitor concentration, v, Km, and Vmax are the velocity, Michaelis-Menten constant, and maximum enzyme velocity without inhibitors, respectively, and v' and Km,app are the velocity and the apparent Km with inhibitors, respectively.
(6)where S is the MDZ concentration, I is the inhibitor concentration, v, Km, and Vmax are the velocity, Michaelis-Menten constant, and maximum enzyme velocity without inhibitors, respectively, and v' and Km,app are the velocity and the apparent Km with inhibitors, respectively.
Cynomolgus Monkey Hepatocyte Uptake Inhibition Assays.
The procedure for the hepatocyte uptake inhibition assay has been described previously (Imaoka et al., 2013). In brief, cryopreserved cynomolgus monkey hepatocytes (Lot DQA, pool of three male monkeys) were thawed using Hepatocyte Thaw Media (Gibco; Thermo Fisher Scientific, Waltham, MA) and resuspended to 2 × 106 viable cells/ml in 37°C prewarmed assay buffer. After warming the cell suspension at 37°C for 3 minutes, the assays were then initiated by the addition of an equal volume of assay buffer containing [3H]RSV with or without curcumin, CG, and CS (final cell concentration: 1 × 106 cells/ml; final test compound concentration, 0.3–30 μM). Simultaneously, to ensure that OATP uptake inhibition occurred properly in the assay, rifampicin was evaluated as a potent inhibitor (final concentration, 100 μM). After incubation at 37°C (0.5 and 1.5 minutes), 60-μl aliquots of the cell mixtures were collected, and then placed in a centrifuge tube containing 100 μl oil (density, 1.015, a mixture of silicone-mineral oil; Sigma-Aldrich) on top of 100 μl 3 M potassium hydroxide solution to separate the cells from the transport buffer. The living cells were passed through the silicone-mineral oil layer by centrifugation at 10,000g for 10 second using a tabletop centrifuge (Beckman Microfuge E; Beckman Coulter, Brea, CA), and then dissolved in a potassium hydroxide solution overnight at room temperature. The radioactivity in both cells and media was determined using a liquid scintillation counter after mixing with a scintillation cocktail.
To confirm the uptake of SASP, 100 μl 5 M ammonium acetate containing the internal standard was used for the living cell separation instead of 3 M potassium hydroxide solution. The sample tubes were centrifuged and stored at −80°C until quantification. An aliquot was taken from the upper media portion and quenched in methanol. The cells were deactivated by sonication and mixing with methanol after being transferred from the centrifuge tube to a new tube. The samples from both the media and cell portions were measured by LC-MS/MS.
The incubations were performed in triplicate. The intrinsic clearance in the hepatocyte uptake assay (CLint, hepatocyte uptake) (microliters per 106 cells/min) was determined from the calculation of the slope of the mean value of triplicate uptake volume (Vd) (microliters per 106 cells) between 0.5 and 1.5 minutes using eq. (7):
 (7)
(7)Plasma Protein Binding Assays in Cynomolgus Monkeys and Humans.
The protein binding [PB (%)] and unbound fraction in plasma (fu) of curcumin, CG, and CS were evaluated in the plasma of cynomolgus monkeys (obtained upon pretreatment after at least a 2-week drug washout period; male, n = 3, pooled) using heparin sodium as an anticoagulant, and of humans (female, n = 3, pooled; purchased from Biopredic International, Inc., Saint Grégroire, France) using EDTA dipotassium as an anticoagulant, processed according to a previously described ultracentrifugation method (Nakai et al., 2004; Srikanth et al., 2013). Briefly, each pooled plasma sample was added to the test substances (total volume: 900 μl; final concentration: 5 μM) and incubated at 37°C for 10 minutes. After incubation, 240 μl of the plasma was transferred into a polycarbonate centrifuge tube in triplicate (Beckman Coulter, Inc.). A 20-μl aliquot was then placed in a tube for measurement of total plasma concentration (Ct). Residual plasma was ultracentrifuged at 4°C and 436,000g for 280 minutes (Optima L-100XP; Beckman Coulter, Inc.) to separate it into three layers according to density. After the upper layer, containing chylomicrons and very low density lipoproteins, was removed by cutting the tube with a tube slicer (Beckman Coulter, Inc.), a 40-μl aliquot of the upper part of the middle layer was obtained as the protein-free fraction for the measurement of the unbound concentration (Cu). All samples were analyzed by LC-MS/MS. The fu and PB (%) were calculated using eqs. (8) and (9):
 (8)
(8) (9)
(9)Analytical Methods.
All sample preparations were performed using protein precipitation by acetonitrile and/or methanol. After mixing, the precipitant was removed by filtration using a Captiva 96-well filter plate (Agilent Technologies, Santa Clara, CA). The resulting supernatants were injected into the LC-MS/MS system consisting of an API 4000 (AB SCIEX, Framingham, MA) and an ACQUITY UPLC system (Waters Corporation, Milford, MA). Both mobile phases were composed of a two-solvent pair: A, 5 mM ammonium acetate and 0.2% formic acid with 5% acetonitrile, and B, 5 mM ammonium acetate and 0.2% formic acid with 95% acetonitrile; C, 5 mM ammonium acetate with 5% acetonitrile, and D, 5 mM ammonium acetate with 95% acetonitrile. The elution gradient for the analysis of RSV, FEX, TLN, curcumin, CG, LAP, PAN, and CS was as follows: solvent B was held at 0.1% for 0.10 minutes, linearly ramped from 0.1 to 50% in 0.10 minutes, ramped from 50.0 to 99.9% in 0.40 minutes, maintained at 99.9% from 0.60 to 0.90 minutes, and then immediately returned to 0.1% for re-equilibration. The gradient for the analysis of MDZ, 1′-hydroxymidazolam, and 4′-hydroxymidazolam was as follows: solvent B was linearly ramped from 0.1 to 20.0% in 0.50 minutes, ramped from 20.0 to 99.9% in 1.15 minutes, maintained at 99.9% for 0.05 minutes, and then immediately returned to 0.1% for re-equilibration. The gradient for the analysis of AL was as follows: solvent D was linearly ramped from 5 to 50% in 0.20 minutes, ramped from 50 to 95% in 0.40 minutes, maintained at 95% from 0.60 to 0.90 minutes, and then immediately returned to 5% for re-equilibration. The gradient for SASP analysis was as follows: solvent D was linearly ramped from 0.1 to 20.0% in 0.20 minutes, ramped from 20.0 to 99.9% in 1.45 minutes, maintained at 99.9% for 0.05 minutes, and then immediately returned to 0.1% for re-equilibration. The gradient for MDZ, 1′-hydroxymidazolam, and 4′-hydroxymidazolam analysis was the same as that for SASP but with substitution with solvent pair A and B. All analytes were chromatographically separated by an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 mm, ID × 50 mm; Waters Corporation) at 50°C with a flow rate of 0.8 ml/min. LC-MS/MS analysis was conducted with multiple reaction monitoring transitions for each test compound using Analyst (version 1.5.1; AB SCIEX) to process the chromatographic data. The precursor and product ion (m/z) pairs of RSV, FEX, TLN, AL, MDZ, curcumin, CG, LAP, PAN, 1′-hydroxymidazolam, 4′-hydroxymidazolam, CS, and SASP were 482.5/258.5, 502.3/466.4, 364.3/308.0, 552.4/436.7, 325.9/290.7, 369.2/177.5, 545.0/369.3, 581.20/365.3, 384.0/200.1, 342.0/203.3, 341.9/234.0 (positive ion mode), 446.4/367.1, and 397.2/197.3 (negative ion mode), respectively. The ion chromatograms were integrated and quantified on the basis of analyte/internal standard peak-area ratios using Analyst. Warfarin was used as the internal standard for the analysis.
Data Analysis
Pharmacokinetics.
The PK parameters [AUCall and the elimination terminal half-life (T1/2) for IV and PO, the maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) for PO, and the apparent volume of disposition at equilibrium (Vdss) for IV] were estimated by noncompartmental analysis using Phoenix WinNonlin. Total body clearance on the basis of plasma concentration (Cltot) was calculated from the dose divided by AUCall after IV administration. T1/2 was calculated from ln2/λz, where λz is the elimination rate constant (the slope of the regression line of several points in the elimination phase as determined by the least squares method). If the adjusted square of the correlation coefficient was less than 0.75, T1/2 was not calculated and was expressed as NC (not calculated) owing to low reliability for fitting. In these cases, Vdss were also expressed as NC. Oral bioavailability (F) was calculated using oral AUCall data and arithmetic mean AUCall after IV administration. The ratios of each PK parameter were calculated (pretreated/untreated).
The Half Maximal Inhibitory Concentration Determination.
The half-maximal (50%) inhibitory concentration (IC50) was determined by fitting the data of each RA or intrinsic uptake clearance to an Inhibitory Imax Model with Phoenix WinNonlin. (10)where C is the concentration of test substances, Ei, E0, and Imax are the activity (the RA or the intrinsic uptake clearance) of each substrate measured at the given inhibitor concentration of test substances, the activity without inhibitor, and the activity of each substrate caused by the maximum inhibition with test substance subtracted from E0, respectively.
(10)where C is the concentration of test substances, Ei, E0, and Imax are the activity (the RA or the intrinsic uptake clearance) of each substrate measured at the given inhibitor concentration of test substances, the activity without inhibitor, and the activity of each substrate caused by the maximum inhibition with test substance subtracted from E0, respectively.
Statistics
All statistical analyses were performed using SAS version 9.2 (SAS Institute, Inc. Cary, NC). Statistical significance was assessed by the paired t test or Shirley-Williams test for PK parameters in the monkey in vivo study. A P value <0.05 was considered statistically significant.
Results
Impact of Curcumin, Lapatinib, and Pantoprazole Pretreatment on Pharmacokinetics of BCRP Substrates in Cynomolgus Monkeys.
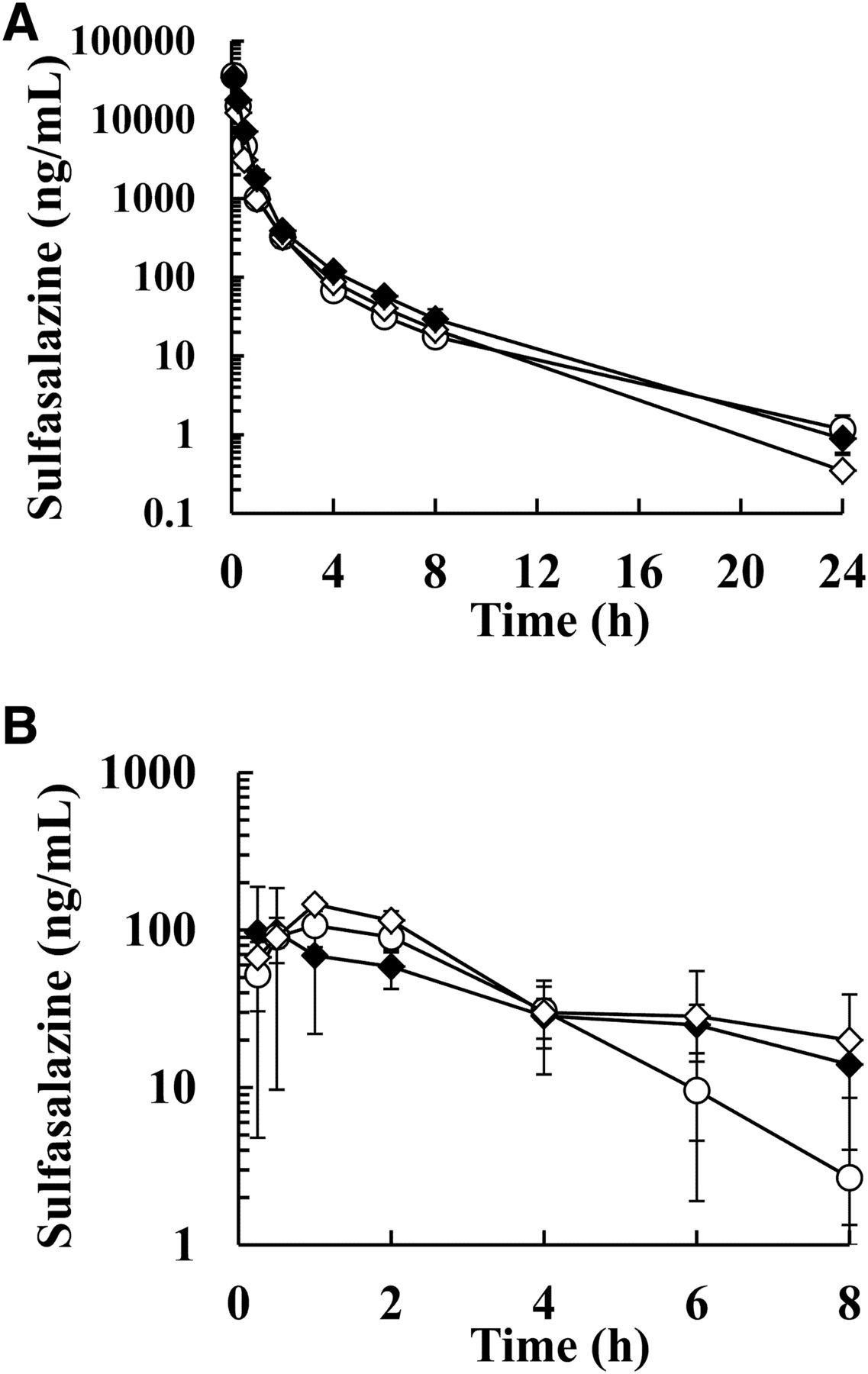
To confirm the inhibition potency of gastrointestinal BCRP function by curcumin, LAP, and PAN, PK studies in cynomolgus monkeys were performed using BCRP substrates (SASP and RSV). The plasma concentrations after a single IV dose of SASP and RSV were comparable in the presence and absence of curcumin pretreatment (Fig. 1, A and C), such that the CLtot changes [curcumin/untreated (UT)] of SASP and RSV were within 1 ± 0.1-fold (Table 2). The plasma concentrations after PO administration of SASP and RSV were higher with than without curcumin pretreatment (Fig. 1, B and D). The oral AUCall and Cmax of SASP were significantly increased by curcumin pretreatment (2.9- and 4.1-fold, respectively; Table 2). A remarkable F increase was also observed after curcumin treatment (3.0-fold). The oral AUCall of RSV after curcumin pretreatment was clearly elevated 1.7-fold compared with that of UT. Curcumin significantly raised the Cmax and F of RSV (2.5- and 1.7-fold, respectively). The Tmax and T1/2 changes of SASP and RSV were slightly less than 2-fold. The PK profiles after a single IV administration of SASP with LAP (5 mg/kg) or PAN (0.6 and 20 mg/kg) were also comparable with that of UT (Figs. 4A and 5A), as CLtot and Vdss for SASP with LAP or PAN were within 1 ± 0.2-fold of those of UT (Tables 3 and 4). In contrast, the mean plasma concentrations of SASP with LAP were higher than those of UT after PO administration (Fig. 4B), whereas those treated with PAN were only higher after 6 hours (Fig. 5B). As shown in Table 3, LAP increased the AUCall, Cmax, and F of SASP 7.5-, 5.9-, and 6.0-fold, respectively. The Tmax of SASP with LAP was comparable to that of UT, although T1/2 of SASP was prolonged after PO administration (3.2 ± 2.0-fold). As summarized in Table 4, the AUCall of SASP at 0.6 and 20 mg/kg PAN was elevated 1.7- and 1.6-fold, respectively, and the Cmax of SASP at 0.6 and 20 mg/kg PAN increased 1.5- and 1.9-fold, respectively. The F of SASP at 0.6 and 20 mg/kg PAN increased 1.8- and 1.4-fold, respectively. Thus, the AUCall, Cmax, and F ratios (PAN/UT) after PO administration of SASP tended to increase, but the changes were smaller than those caused by curcumin and LAP pretreatment. The Tmax slightly changed at 20 mg/kg of PAN (2.3-fold) but was comparable with that of UT at 0.6 mg/kg (0.88-fold).

Plasma concentration profiles after single intravenous and oral administrations of breast cancer resistance protein substrates (sulfasalazine and rosuvastatin) in the presence (30 mg/kg: ●) or absence (○) of curcumin in cynomolgus monkeys. (A) Sulfasalazine (IV, 5 mg/kg, N = 9), (B) sulfasalazine (PO, 5 mg/kg, N = 10), (C) rosuvastatin (IV, 1 mg/kg, N = 6), and (D) rosuvastatin (PO, 1 mg/kg, N = 6). Each point represents the mean ± S.E. of each group.
Pharmacokinetic parameters after single intravenous and oral administrations of breast cancer resistance protein substrates (sulfasalazine and rosuvastatin) to cynomolgus monkeys with or without curcumin pretreatment
Data are expressed as means ± S.E. of cynomolgus monkeys in each group. There were no significant differences between UT and curcumin pretreatment after single IV injection as determined by a paired t test.
Pharmacokinetic parameters after single intravenous and oral administrations of sulfasalazine, fexofenadine, and midazolam to cynomolgus monkeys with or without lapatinib pretreatment
Data are expressed as means ± S.E. of cynomolgus monkeys in each group. There were no significant differences between UT and LAP pretreatment after single IV injection of SASP and MDZ as determined by a paired t test.
Pharmacokinetic parameters after single intravenous and oral administrations of sulfasalazine in cynomolgus monkeys with or without pantoprazole pretreatment
Data are expressed as means ± S.E. of cynomolgus monkeys in each group. There were no significant differences between UT and PAN pretreatment as measured by a Shirley-Williams test.
The monkey PK profiles of inhibitors used in this study were as follows: With curcumin pretreatment, only CG could be monitored through the plasma PK profile [mean Cmax: 117 ± 12 ng/ml (0.215 ± 22 μM), Tmax: 4.02 ± 0.36 hours], whereas the plasma concentrations of curcumin and CS in almost all samples measured in these studies were below the lower limit of quantitation (curcumin: 10 ng/ml; CS: 100 ng/ml). The mean Cmax and Tmax of LAP after pretreatment were 80.0 ± 11.8 ng/ml (0.138 ± 0.020 μM) and 3.06 ± 0.36 hours, respectively. The mean Cmax of PAN after pretreatment at 0.6 and 20 mg/kg was 340 ± 103 (0.887 ± 0.269 μM) and 15,300 ± 5900 ng/ml (39.9 ± 15.4 μM), respectively, whereas the mean Tmax of PAN after pretreatment at 0.6 and 20 mg/kg was 0.920 ± 0.327 and 1.21 ± 0.56 hours, respectively.
Impact of Curcumin or Lapatinib Pretreatment on Pharmacokinetics of P-gp Substrates in Cynomolgus Monkeys.
To understand the effect of curcumin and LAP on intestinal efflux transport via P-gp, monkey PKs after a single PO administration of P-gp substrates (FEX, TLN, and AL) were evaluated. The plasma concentrations of FEX, TLN, and AL with curcumin were comparable to those without curcumin, although these exposures were elevated by pretreatment with 5 mg/kg EL, a potent P-gp inhibitor (Fig. 2). The AUCall of FEX, TLN, and AL with curcumin were comparable to those without (1.0-, 1.0-, and 1.1-fold, respectively), whereas it significantly increased 5.7-, 1.6-, and 4.0-fold, respectively, with EL pretreatment compared with UT (Table 5). The Cmax of FEX, TLN, and AL was not significantly changed (1.3-, 0.90-, and 1.4-fold, respectively), whereas it increased with EL pretreatment compared with UT (11-, 1.6-, and 6.4-fold, respectively). The Tmax and T1/2 differences between FEX and TLN were slightly less than 2-fold for both treatments. The Tmax ratios of AL after curcumin and EL pretreatment varied widely (4.5 ± 3.8- and 6.6 ± 5.8-fold, respectively). The T1/2 change in AL was not evaluated, because T1/2 with UT could not be estimated owing to the low reliability for fitting. In contrast to curcumin, higher plasma concentrations of FEX with LAP were observed for up to 4 hours after a single PO administration of FEX than those observed in the UT group (Fig. 4C). The AUCall and Cmax of FEX with LAP were clearly elevated 2.4- and 5.1-fold, respectively (Table 3). The Tmax of FEX with LAP showed no significant change, although a shorter T1/2 of FEX was observed with LAP than with UT (0.80-fold).

Plasma concentration profiles after single oral administration of P-glycoprotein substrates (fexofenadine, talinolol, and aliskiren) with curcumin (30 mg/kg: ●), elacridar (5 mg/kg: □), or untreated (○) in cynomolgus monkeys. (A) Fexofenadine (PO, 2 mg/kg, N = 7), (B) talinolol (PO, 1 mg/kg, N = 4), and (C) aliskiren (PO, 3 mg/kg, N = 4). Each point represents the mean ± S.E. of each group.
Pharmacokinetic parameters after single oral administrations of P-glycoprotein substrates (fexofenadine, talinolol, and aliskiren) to cynomolgus monkeys with or without curcumin or elacridar pretreatment
Data are expressed as means ± S.E. of cynomolgus monkeys in each group. There were no significant differences, except in Tmax, between UT and curcumin pretreatment as determined by a paired t test.
Impact of Curcumin and Lapatinib Pretreatment on Pharmacokinetics of Midazolam in Cynomolgus Monkeys.
To investigate the impact of curcumin and LAP on intestinal and hepatic CYP3A activity, MDZ was used as an in vivo CYP3A probe for monkeys. The PK profiles after a single IV administration were similar with or without pretreatment with curcumin and KTZ as in vivo intestinal CYP3A inhibitors (Fig. 3A), as their CLtot changes were within 1 ± 0.2-fold (Table 6). In contrast, the mean plasma concentrations of MDZ were increased after PO administration with curcumin and KTZ pretreatment compared with those of UT (Fig. 3B). The AUCall of MDZ with curcumin and KTZ was significantly elevated (1.4- and 7.3-fold, respectively). Curcumin and KTZ raised the Cmax (1.1- and 9.5-fold, respectively) and the F (1.6- and 8.2-fold, respectively). The changes in Tmax and T1/2 after curcumin pretreatment were prolonged 2.0-and 4.1-fold, respectively, although the values after KTZ pretreatment were comparable with those after UT. Meanwhile, the differences in CLtot and Vdss between the UT and LAP treatments were less than ±10% after IV dosing with MDZ, whereas a marginally significant increase in AUCall after LAP treatment was observed for unknown reasons compared with that after UT (1.1-fold; Table 3). The plasma concentrations after a single PO administration of MDZ were also higher after LAP pretreatment than with UT (Fig. 4E). LAP raised the oral AUCall and Cmax of MDZ compared with UT (both 1.8-fold, Table 3). LAP pretreatment increased the F of MDZ 1.6-fold. The Tmax of MDZ showed no significant change.

Plasma concentration profiles after single intravenous and oral administrations of midazolam with curcumin (30 mg/kg: ●), ketoconazole (4 mg/kg: ▪), or untreated (○) in cynomolgus monkeys. (A) Intravenous administration (0.2 mg/kg, N = 6) and (B) oral administration (2 mg/kg, N = 8). Each point represents the mean ± S.E. of each group.
Pharmacokinetic parameters after single intravenous and oral administrations of midazolam in cynomolgus monkeys with or without curcumin or ketoconazole pretreatment
Data are expressed as means ± S.E. in each group. There were no significant differences between UT and curcumin or ketoconazole pretreatment after midazolam IV as determined by a paired t test.

Plasma concentration profiles after single intravenous and oral administrations of sulfasalazine, fexofenadine, and midazolam with lapatinib (5 mg/kg: Δ) or untreated (○) in cynomolgus monkeys. (A) Sulfasalazine (IV, 5 mg/kg, N = 3), (B) sulfasalazine (PO, 5 mg/kg, N = 4), (C) fexofenadine (PO, 2 mg/kg, N = 4), (D) midazolam (IV, 0.2 mg/kg, N = 3), and (E) midazolam (PO, 2 mg/kg, N = 4). Each point represents the mean ± S.E. in each group.

Plasma concentration profiles after single intravenous and oral administrations of sulfasalazine with pantoprazole (0.6 mg/kg: ⋄, 20 mg/kg: ♦) or untreated (○) in cynomolgus monkeys. (A) Intravenous administration (5 mg/kg, N = 3) and (B) oral administration (5 mg/kg, N = 4). Each point represents mean ± S.E. in each group.
Inhibitory Effect on Efflux Transporters in Caco-2 Cells.
To clarify the inhibition of intestinal efflux transporters by the in vivo inhibitors as observed AUC changes, the IC50 values of curcumin, the main curcumin metabolites (CG and CS), LAP, and PAN in the efflux transport of probes for BCRP (ES, SASP, and RSV) and P-gp (digoxin, FEX, TLN, and AL) were calculated from efflux transporter inhibition assays using Caco-2 cells (Table 7). The Caco-2 efflux of BCRP substrates was inhibited by curcumin, LAP, and PAN in a dose-dependent manner, and also by novobiocin (a potent inhibitor of BCRP), but not by verapamil (a strong inhibitor of P-gp). In contrast, the Caco-2 efflux of P-gp substrates was weak or not observed within 100 μM after treatment with curcumin, PAN, or novobiocin, and was blocked by LAP and verapamil (data not shown for novobiocin and verapamil). The Caco-2 permeability of MDZ was not influenced by curcumin, LAP, novobiocin, or verapamil, indicating that MDZ PK were not affected by BCRP/P-gp inhibition. The IC50 values of curcumin for ES, SASP, and RSV were 8.23, 17.4, and 9.55 μM, respectively, whereas the IC50 values of curcumin for FEX, TLN, and AL were more than 100 μM, and the IC50 for digoxin was 56.2 μM. The IC50 values of LAP for ES, SASP, and RSV were 0.0567, 0.0431, and 0.256 μM, respectively, whereas the IC50 values of LAP for digoxin, FEX, and TLN were 2.47, 4.01, and 2.65 μM, respectively. The IC50 values of PAN for ES, SASP, RSV, and digoxin were 4.42, 3.22, 14.1, and 55.9 μM, respectively. For CG and CS, the IC50 values for ES and digoxin were more than 100 μM. RSV efflux transport was inhibited by CG and CS (IC50: 5.70 and 13.8 μM, respectively) although the IC50 value for SASP was more than 100 μM.
IC50 values (μM) of curcumin, curcumin glucuronide, curcumin sulfate, lapatinib, and pantoprazole for efflux transporter substrates in Caco-2 cells
Inhibitory Effect on Midazolam Hydroxylation in Liver and Small Intestinal Microsomes of Cynomolgus Monkeys and Humans.
To elucidate the cause of AUC increase of MDZ by curcumin pretreatment, the CYP3A inhibition potentials for MDZ 1′-hydroxylation and 4′-hydroxylation were evaluated using liver and small intestinal microsomes from cynomolgus monkeys and humans. No inhibition of MDZ 1′-hydroxylation and 4′-hydroxylation was observed with treatment of 0.5–50 μM CG; in contrast, curcumin and CS showed less than 50% inhibition compared with that of UT (data not shown). Therefore, the Ki of curcumin and CS for MDZ 1′-hydroxylation and 4′-hydroxylation were estimated by the Km,app method. As summarized in Table 8, the Ki values of curcumin for midazolam 1′-hydroxylation and 4′-hydroxylation were comparable between monkeys and humans, regardless of liver or small intestinal microsomes. Conversely, the Ki value of CS for 1′-hydroxylation (46.6 μM) was different from that for 4′-hydroxylation (14.3 μM) in cynomolgus monkey small intestinal microsomes, although the Ki values of CS for 1′-hydroxylation and 4′-hydroxylation were comparable in other microsomes tested (17.0 and 19.5 μM in cynomolgus monkey liver microsomes, 3.20 and 6.95 μM in human liver microsomes, and 4.54 and 7.26 μM in human small intestinal microsomes, respectively).
Summary of estimates of Km, Vmax, and Ki for midazolam 1′-hydroxylation and 4′-hydroxylation in the liver and the small intestinal microsomes of cynomolgus monkeys and humans by Km, app method
Inhibitory Effect on Uptake into Cynomolgus Monkey Hepatocytes.
To estimate the possibility of AUC increase of BCRP substrates via OATP inhibition by curcumin pretreatment, as curcumin and CG have strong inhibition potentials for human OATP1B1 and OATP1B3 (Sun et al., 2016; Zhou et al., 2017), the IC50 values of curcumin, CG, and CS in the hepatic uptake of BCRP substrates (SASP and RSV) were calculated by uptake transporter inhibition assays using cryopreserved cynomolgus monkey hepatocytes. Plasma protein binding was also evaluated. Hepatic uptake of BCRP substrates was inhibited by all compounds tested in a dose-dependent manner, and was also reduced by rifampicin, a potent inhibitor of OATPs (data not shown). The IC50 values of curcumin, CG, and CS for SASP were 2.46, 5.73, and 0.343 μM, respectively, whereas the values for RSV were 4.82, 4.95, and 1.86 μM, respectively.
The in vitro plasma protein binding levels of curcumin, CG, and CS were very high in cynomolgus monkeys and humans. The monkey plasma protein binding for curcumin, CG, and CS was 99.7%, 99.2%, and 99.6%, respectively; likewise, the human plasma protein binding for curcumin, CG, and CS was 99.8%, 99.6%, and 99.6%, respectively.
Discussion
This study aimed to confirm the appropriateness of curcumin, an in vivo clinical BCRP inhibitor, in the inhibition of gastrointestinal BCRP function and to assess the impact on intestinal and hepatic uptake, P-gp, and CYP3A activity in cynomolgus monkeys. Using well known substrates of BCRP (SASP and RSV), P-gp (FEX, TLN, and AL), and CYP3A (MDZ), exposure changes caused by curcumin were evaluated in comparison with those caused by LAP and PAN. To investigate the factors affecting AUC changes observed in vivo, in vitro plasma protein binding assays as well as inhibition potential studies of BCRP, P-gp, CYP3A, and hepatic uptake transporters were performed.
For BCRP, the oral AUCall ratios of SASP and RSV were significantly increased by curcumin pretreatment in monkeys (SASP, 2.9 ± 0.4; RSV, 1.7 ± 0.2) without affecting systemic clearance. The AUCall ratios with curcumin were comparable to geometric mean AUC ratios in human ABCG2 421AA/CC subjects (SASP: 2.1–3.5; RSV: 1.6–3.2) as well as those after EL pretreatment (SASP, 6.1 ± 1.9; RSV, 3.1 ± 1.8; (Karibe et al., 2015)). Neither LAP nor PAN affected systemic clearance of SASP. The oral AUCall ratio for SASP with LAP (7.5 ± 2.4) was elevated more than that for SASP with curcumin, although one nonresponder was observed (Table 3); on the other hand, the ratios for SASP with PAN (maximum 1.7 ± 0.3 at 0.6 mg/kg) were increased dose independently, although the increase was not as significant as previously observed by Adkison et al. (2010) and was less than that caused by curcumin. Thus, curcumin and LAP, like EL, were useful in vivo inhibitors for gastrointestinal BCRP assessment.
For P-gp substrates (FEX, TLN, and AL) administered orally, curcumin caused few changes in plasma concentrations and AUCall (within 1 ± 0.1-fold), whereas EL significantly increased AUCall compared with UT, validating the intestinal P-gp function model in monkeys. LAP also remarkably elevated oral FEX AUCall; therefore, curcumin selectively inhibited gastrointestinal BCRP function without affecting P-gp-mediated efflux transport.
Considering in vitro efflux inhibition in Caco-2 cells, the selected doses of curcumin (30 mg/kg), LAP (5 mg/kg), and PAN (0.6 and 20 mg/kg) might have sufficient potential to exert an AUC increase by BCRP inhibition in monkeys because the estimated maximum inhibitor concentrations in the gut [dose/gut volume in cynomolgus monkeys (46 ml/kg; Peters, 2012); curcumin 1770 μM, LAP 187 μM, and PAN ≥34.0 μM] were much higher than the IC50 values for BCRP (Table 7). In line with in vivo observations, LAP appeared to show the same potential for AUC increase of FEX by P-gp inhibition as it did by BCRP inhibition, whereas curcumin inhibition potency for FEX, TLN, and AL was weaker (>100 μM). Intestinal concentration of curcumin and LAP in monkeys may be overestimated owing to very poor solubility (Sasaki et al., 2011; https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/022059s022lbl.pdf), unlike PAN, which is water-soluble and readily absorbed from the gut (https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020987s050,022020s012lbl.pdf). Thus, the actual intestinal concentration of curcumin in monkeys might lead to higher IC50 values for BCRP but not P-gp substrates, as indicated by its solubility at pH 7.0 (approximately 24 μM, in-house data). On the other hand, the actual intestinal concentration of LAP was presumably higher than the IC50 values for FEX and SASP, as its water-solubility is 0.007 mg/ml (12 μM; https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/022059s022lbl.pdf). Further studies to estimate gastrointestinal concentration transitions in poorly water-soluble drugs are required to understand DDIs in the gut.
For CYP3A, curcumin and LAP significantly elevated the AUCall and F ratio of MDZ after PO administration with minimal impact on systemic clearance, which was attributable to intestinal CYP3A inhibition. In vivo inhibition potency for intestinal CYP3A activity by curcumin was comparable with that by LAP, as shown by the similar AUCall and F ratios of MDZ after curcumin and LAP treatments (Tables 6 and 3). Collectively, curcumin is a more appropriate BCRP inhibitor for the evaluation of absorptional BCRP contribution than LAP and PAN are, with significant in vivo BCRP inhibition, limited effects on P-gp substrate PK, and inhibition potency for intestinal CYP3A comparable to that of LAP.
In addition to curcumin, CG and CS were assessed for their inhibition potential as causes of oral AUC increase for BCRP substrates and MDZ by curcumin pretreatment, because they were detected in plasma after oral curcumin administration to humans despite low plasma concentrations of curcumin itself (Garcea et al., 2004; Vareed et al., 2008). In the present study, only CG could be monitored in monkey plasma. The SASP AUC increase resulting from BCRP inhibition by CG and CS was considered low, because the IC50 value for SASP in Caco-2 cells could not be estimated by CG or was relatively high by CS. In contrast, CG and CS were able to elevate the RSV AUC owing to their comparable IC50 values for the BCRP-mediated efflux of RSV (curcumin, CG, CS: 9.55, 5.70, 13.8 μM, respectively). With regard to CYP3A activity, CG showed no inhibition of MDZ hydroxylation in human or monkey liver and small intestinal microsomes, whereas curcumin and CS showed inhibition in both microsomes with similar Ki values (Table 8). CYP3A inhibition was considered to be caused by curcumin and CS in the gut rather than the liver owing to unchanged systemic clearance regardless of curcumin pretreatment, the absence of plasma concentrations of curcumin and CS, and high plasma protein binding of curcumin and CS. Ogasawara et al. (2007) reported that a significant increase in oral MDZ exposure was confirmed by KTZ with limited changes in systemic clearance, suggesting that KTZ mainly inhibits intestinal CYP3A activity in monkeys. At least ∼80% of intestinal CYP3A activity remained after curcumin pretreatment compared with the F ratio after KTZ treatment (KTZ/UT). However, the Ki values of curcumin (Table 8) in monkey small intestinal microsomes were lower than the IC50 values for BCRP substrates in Caco-2 cells (Table 7). This difference in the impact on PK between CYP3A and BCRP can presumably be attributed to the site of inhibition; BCRP was inhibited in both intracellular and extracellular sites in the gut, whereas intracellular concentration was important for cytochrome P450 inhibition. Therefore, low solubility and permeability of curcumin caused partial intestinal CYP3A inhibition, although its intestinal concentration was sufficient to demonstrate BCRP inhibition.
With regard to the AUC increases of SASP and RSV by inhibition of hepatocyte uptake transporters, the potency after oral curcumin pretreatment was improbable, because systemic clearances of SASP and RSV were comparable regardless of curcumin pretreatment, despite observations of the uptake inhibition for SASP and RSV in monkey hepatocytes from curcumin, CG, and CS. Zhou et al. (2017) reported that curcumin coadministration at 500 and 100 mg/kg significantly increased RSV exposure in rats and dogs, respectively, which was considered to result from OATP inhibition without the evaluation of systemic clearance changes of RSV with curcumin. However, the plasma concentration of CG in their study (164 ± 22 ng/ml (0.301 ± 0.040 μM] at curcumin 100 mg/kg in rats) was presumed to be insufficient for the inhibition of hepatic OATPs, because the estimated unbound Cmax of CG was lower than the IC50 values for human OATPs in OATP1B1- and OATP1B3-transfected HEK-293 cells (1.04 and 1.08 μM, respectively; Zhou et al., 2017) when high-plasma protein bindings in rats and dogs were expected (>99%) from our findings. Moreover, oral exposure of FEX, which has been associated with OATP1B1 polymorphism (Niemi et al., 2005), showed minimal change with curcumin in monkeys (Table 5). These results indicated that inhibition of hepatic uptake transporters for SASP and RSV by curcumin and its metabolites resulted in limited PK effects.
In the case of intestinal uptake transporters (OATP1A2 and OATP2B1), curcumin shows an inhibition potential for OATP2B1 (Kusuhara et al., 2012; Zhou et al., 2017), which is considered to contribute to SASP and RSV uptake in the gut. However, significant reductions in oral AUCall and Cmax for FEX, TLN, and AL were not observed after curcumin administration (Table 5), although these compounds, as well as P-gp, were also known to be OATP1A2 and/or OATP2B1 substrates (Bailey, 2010; Tapaninen et al., 2011; Rebello et al., 2012; Akamine et al., 2015). These results implied that OATP1A2 and OATP2B1 inhibition after curcumin pretreatment was limited in monkeys or that the saturation of those transporters was observed under these study conditions.
In conclusion, PK evaluation in monkeys after pretreatment with curcumin as an in vivo BCRP inhibitor is more appropriate for the assessment of BCRP impact on gastrointestinal absorption in a nonrodent model than is pretreatment with LAP or PAN. Curcumin resulted in minimal PK changes to hepatic uptake transporters, P-gp, and weak inhibition of intestinal CYP3A activity. For non-CYP3A substrates, our approach can be adopted immediately. For CYP3A substrates, curcumin is effective in combination with KTZ pretreatment to assess BCRP contribution to intestinal absorption separately from the impact of intestinal CYP3A activity on monkey PK. As the clinical impact of BCRP on drug disposition is much more pronounced for orally than for intravenously administered drugs, this study contributes not only to a more adequate estimation of the impact of BCRP modulation on investigational drug PK in humans than is possible using EL, but also to the advancement of clinical DDI studies via BCRP with curcumin pretreatment.
Acknowledgments
The authors thank Ken-ichi Itokawa in Drug Metabolism and Pharmacokinetics Research Laboratories, Daiichi Sankyo Co., Ltd. and the staff and researchers of Narita Animal Sciences for technical assistance.
Authorship Contributions
Participated in research design: Karibe, Imaoka, Abe.
Conducted experiments: Karibe.
Performed data analysis: Karibe.
Wrote or contributed to the writing of the manuscript: Karibe, Imaoka, Abe, Ando.
Footnotes
- Received October 12, 2017.
- Accepted January 19, 2018.
This study was supported by Daiichi Sankyo Co., Ltd.
Abbreviations
- AL
- aliskiren
- AUC
- area under the plasma concentration-time curve
- AUCall
- AUC from 0 h to the time of the last observation
- BCRP
- breast cancer resistance protein
- CG
- curcumin β-d-glucuronide
- CLtot
- total body clearance on the basis of plasma concentration
- Cmax
- the maximum plasma concentration
- CS
- curcumin sulfate
- DDI
- drug-drug interaction
- DMSO
- dimethyl sulfoxide
- EL
- elacridar
- ES
- estrone sulfate
- F
- bioavailability
- FEX
- fexofenadine
- IC50
- half maximal (50%) inhibitory concentration
- IV
- intravenous, intravenously
- Ki
- the inhibition constant
- Km
- the velocity, the Michaelis-Menten constant
- KTZ
- ketoconazole
- LAP
- lapatinib
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MC
- aqueous methylcellulose
- MDZ
- midazolam
- OATP
- organic anion-transporting polypeptide
- PAN
- pantoprazole
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- PO
- by mouth
- RSV
- rosuvastatin
- SASP
- sulfasalazine
- T1/2
- the elimination terminal half-life
- TLN
- talinolol
- Tmax
- the time to reach Cmax
- Vdss
- apparent volume of disposition at equilibrium
- UT
- untreated
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}