


Abstract
The mechanism underlying subcutaneous absorption of macromolecules and factors that can influence this process were studied in rats using PEGylated erythropoietins (EPOs) as model compounds. Using a thoracic lymph duct cannulation (LDC) model, we showed that PEGylated EPO was absorbed from the subcutaneous injection site mainly via the lymphatic system in rats, which is similar to previous reports in sheep. After subcutaneous administration, the serum exposure was reduced by ∼70% in LDC animals compared with that in the control animals, and most of the systemically available dose was recovered in the lymph. In both LDC and intact rats, the total radioactivity recoveries in excreta after subcutaneous administration were high (70–80%), indicating that catabolism, not poor absorption, was the main cause for the observed low bioavailability (30–40%). Moreover, catabolism of PEGylated EPO was found with both rat subcutaneous tissue homogenate and lymph node cell suspensions, and a significant amount of dose-related breakdown fragments was found in the lymph of LDC rats. In addition, the bioavailability of PEGylated EPOs was shown to be 2- to 4-fold lower in “fat rats,” indicating that physiologic features pertinent to lymphatic transport can have a profound impact on subcutaneous absorption. Limited studies in dogs also suggested similar subcutaneous absorption mechanisms. Collectively, our results suggest that the lymphatic absorption mechanism for macromolecules is probably conserved among commonly used preclinical species, e.g., rats and dogs, and that mechanistic understanding of the subcutaneous absorption mechanism and associated determinants should be helpful in biologic drug discovery and development.
Introduction
Almost all protein-based biologic drugs are administered parenterally. Subcutaneous administration is the preferred route of administration compared with intravenous administration. There are more than 30 marketed biologic drugs that are administered subcutaneously, including most of the therapeutic proteins/peptides (McDonald et al., 2010). However, despite this wide application, our understanding of the exact mechanism underlying subcutaneous absorption and the factors that can influence this process remain limited (Porter and Charman, 2000; Lin, 2009).
Biologic drugs can exhibit a wide range of bioavailability after subcutaneous administration in humans (Tang et al., 2004). Many physiologic factors, e.g., age, body weight, and injection site, have been identified as covariants for the pharmacokinetics (PK) of macromolecules administered subcutaneously in humans (Macdougall et al., 1991; Chan et al., 2003; Fishbane et al., 2007; Olsson-Gisleskog et al., 2007; Kakkar et al., 2011). However, the type of factors and extent of their impact are apparently compound-dependent. For example, when the PK of peginterferon alfa-2a and peginterferon alfa-2b were compared in a randomized 36-patient trial, peginterferon alfa-2a showed much greater variability in patient exposure than peginterferon alfa-2b (38% versus 20%), and body weight appeared to be a covariant for peginterferon alfa-2a, but not for peginterferon alfa-2b (Silva et al., 2006). At present, there is no established method to predict subcutaneous absorption in humans a priori, i.e., bioavailability, potential covariants, or the magnitude of PK variability. Therefore, there is a great need for more mechanistic understanding of the subcutaneous absorption process, as well as more preclinical and in vitro tools to help gain insights into its underlying mechanism.
Our current understanding of the subcutaneous absorption process mostly came from a sheep lymphatic cannulation model, in which a series of studies demonstrated a molecular weight-dependent lymphatic contribution to the subcutaneous absorption of macromolecules; i.e., macromolecules with molecular mass >16 kDa are absorbed mainly via the lymphatic system (Supersaxo et al., 1990; Porter and Charman, 2000; Porter et al., 2001; McLennan et al., 2006). In addition, the observation of dose-dependent subcutaneous bioavailability for an anti-CD4 monoclonal antibody also provided indirect evidence that monoclonal antibodies are absorbed via the lymphatic pathway in mice (Davis and Bugelski 1998). This theory aligns well with our knowledge of the anatomic structural differences between blood and lymph capillaries. The lymphatic system is a unidirectional system that transports excess fluid from interstitium to systemic circulation. Unlike blood capillaries in the subcutaneous space whose tight endothelial junctions make it difficult for macromolecules to pass through, lymphatic capillaries have incomplete basal lamina, which enables almost unrestricted drainage of macromolecules from the interstitial space (Swartz, 2001). Because of the technical challenges of cannulating lymph vessels in smaller-sized laboratory animals, there have been very limited reports on the contribution of the lymphatic system in subcutaneous absorption of macromolecules in commonly used preclinical species. Unfortunately, the few available reports in rats and rabbits presented contradictory evidence (Bocci et al., 1986, 1988; Kojima et al., 1988; Kagan et al., 2007). In the more recent report by Kagan et al. (2007), the contribution of the lymphatic system to subcutaneous absorption of macromolecules was systematically examined in a rat thoracic lymph duct cannulation (LDC) model, using bovine insulin (5.6 kDa), recombinant human erythropoietin (EPO) (30.4 kDa), and bovine albumin (66 kDa) as model compounds. The results suggested minimal contribution of the lymphatic system for all three molecules, regardless of their molecular masses, i.e., minimal reduction of serum exposure in the thoracic LDC animals, and <3% drug recovery in thoracic duct lymph (Kagan et al., 2007). The only report in which a significant percentage (up to 29%) of subcutaneous dosed radioactivity was recovered in the rat thoracic duct lymph was a study using PEGylated polylysine dendrimers, although the amount of dose recovered in the lymph was still considerably lower than the systemic bioavailability (94–104%) (Kaminskas et al., 2009). These results raised the question of whether the contribution of the lymphatic system in subcutaneous absorption of macromolecules is conserved among preclinical species and challenged the value, if any, of studying subcutaneous absorption in these commonly used preclinical species.
Here, we report our efforts in elucidating the mechanism of subcutaneous absorption of macromolecules in thoracic LDC models in rats and dogs. Two PEGylated recombinant human EPOs were selected as model compounds because they are likely to be absorbed via the lymph, and their PK had been well characterized in rats and dogs. Their catabolism after subcutaneous administration and the impact of physiologic features pertinent to lymphatic absorption/transport, e.g., fat content and level of physical activity, on subcutaneous absorption were also evaluated in these preclinical models.
Materials and Methods
Materials.
Recombinant human EPO is a 30.4-kDa therapeutic protein. PEG30-EPO [continuous erythropoietin receptor activator (CERA)] is a commercially available PEGylated recombinant human EPO that contains a ∼30-kDa methoxy PEG chain linked via amide bonds to the N-terminal amino group or the ε-amino group of lysines (predominantly lysine-52 or lysine-45) (Macdougall, 2005). CERA (Mircera) was purchased from Myoderm (Norristown, PA). PEG40-EPO is a PEGylated recombinant human EPO produced in-house, which has an ∼40-kDa linear PEG conjugated preferably to the amino terminus of the recombinant human EPO (Nett et al., 2012). In addition to the difference in PEG sizes, PEG30-EPO was produced from Chinese hamster ovary cells with enriched tetraantennary sialylated glycoforms at the three N-glycosylation sites of EPO, whereas PEG40-EPO was produced from engineered strains of Pichia pastoris with terminally sialylated biantennary N-glycan structures (Nett et al., 2012).
125I Labeling.
125I labeling of PEG30-EPO and PEG40-EPO was conducted using Iodobeads according to the manufacturer's protocol with minor modification (Thermo Fisher Scientific, Waltham, MA). In brief, 100 μg of protein dialyzed into phosphate-buffered saline was incubated with 1 mCi of 125I (PerkinElmer Life and Analytical Sciences, Waltham, MA) and two Iodobeads for 15 min at room temperature. The labeled protein was purified with a Zeba column (Thermo Fisher Scientific), and stored at 4°C. The radiochemical concentration was determined by gamma counting using a Wallac 1470 automated gamma counter (PerkinElmer Life and Analytical Sciences). The purity of labeled proteins was determined by a size-exclusion high-performance liquid chromatography system equipped with a gamma detector. The percentage of free 125I was less than 2% in all preparations. The integrity of labeled proteins was also analyzed with a product-specific immunoassay (Quantikine IVD human erythropoietin kit; R&D Systems, Minneapolis, MN), and all labeled proteins showed concentration-response curves comparable to those of the corresponding unlabeled materials. For in vivo dosing, the dosing solution was prepared by mixing unlabeled compound, 125I-labeled compound, and formulation buffer to achieve the desired specific activity and final protein concentration.
In Vivo Studies in Sprague-Dawley Rats and Beagle Dogs.
All protocols were approved by the Merck Institutional Animal Care and Use Committee.
Thoracic LDC in rats.
Male Sprague-Dawley (SD) rats (350–400 g; Taconic Farms, Germantown, NY) were used for all rat lymph cannulation studies. Rats were given 0.5 to 1.0 ml of olive oil by oral gavage 0.5 to 1 h before the surgery to provide enhanced visualization of lymph vessels. The thoracic duct cannulation technique was based on previous reports (Lee and Hashim, 1966; Ionac, 2003) with modifications for continuous lymph collection.
In brief, rats were anesthetized with isoflurane. The thoracic duct was located and separated from the psoas muscle and dorsal aorta. The duct was cannulated with a heparin (500 units/ml) saline-filled catheter. All accessory branches into the main lymph duct were either bypassed by the cannula tip or ligated. Heparin (500 units/ml) was also infused at a constant flow rate of 50 μl/h into the catheter that contains collected lymph to prevent the coagulation of lymph fluid ex vivo and therefore maintain the free flow of lymph fluid. Once the lymph flow had been established, the cannula was tied down by silk ligatures tunneled subcutaneously and exteriorized at the back between the shoulder blades. A spring tether system was installed and attached to a swivel to allow free movement for the animals. After the surgical procedure, the rats were monitored until they regained full consciousness. The rats were provided with ad libitum rodent chow and recovery gel (Diet Gel, Clear H2O). There was a 48-h period of recovery and stabilization with continuous lymph fluid collection before initiation of the study.
For in vivo dosing, 125I-labeled PEG30-EPO (25 μg/kg, 13.5 μCi/kg) or PEG40-EPO (36 μg/kg, 14 μCi/kg) was administered subcutaneously to the lateral left lower hind leg (just below the knee) of the thoracic LDC rats as well as the sham-operated rats. Thoracic duct lymph was continuously collected for all cannulated animals during the entire 7-day study period, and the volumes were recorded. The lymph flow ranged from 0.5 to 5 ml/h for LDC rats, and the total volume of lymph collected per animal ranged between 25 and 650 ml during the study period. Only data from animals with total collected lymph volume of >70 ml were used. A series of blood samples were collected for all animals from the carotid artery via implanted cannula at predefined time intervals and serum was prepared. Lymph and serum samples were stored at −70°C until analysis. For the study with PEG30-EPO, the animals were kept in metabolic cages, and urine and feces samples were also collected.
Thoracic LDC in beagle dogs.
Thoracic LDC in beagle dogs was performed similarly to that in SD rats. In brief, adult male beagle dogs were surgically prepared 16 h before dosing. The lymph duct was cannulated close to the thoracic duct ampulla with a Silastic catheter, which was tunneled under the skin to a small incision over the animal's shoulder region and exteriorized. All accessory branches into the main lymph duct were either bypassed by the cannula tip or ligated. After the surgical procedure, the dogs were monitored until they regained full consciousness. An electrolyte mix was administered subcutaneously to the dogs to help compensate for the liquid loss and maintain the lymph flow. The lymph flow after surgery was monitored; it ranged from 1 to 50 ml/h for LDC dogs, and the total volume of lymph collected per animal ranged between 10 and 700 ml during the study period. Only the animals that maintained a stable lymph flow at the time of study initiation were used for the following studies.
For in vivo dosing, 125I-labeled PEG40-EPO (10 μg/kg, 7.5 μCi/kg) was administered either intravenously or subcutaneously (at the lateral left lower hind leg, just below the knee) to thoracic LDC animals. Thoracic duct lymph was continuously collected, and the volumes were recorded. Only data from the two animals with sustained lymph flow for >48 h were used. Blood samples were also collected from the jugular vein at predefined time intervals, and serum was prepared. Lymph and serum samples were stored at −70°C until analysis.
PK Study in Fat Rats to Assess Impact of Fat on Subcutaneous Absorption.
“Fat rats” were obtained by feeding 2- to 3-month-old regular SD rats (∼250 g) with a high-fat diet for ∼3 months. These fat rats had an average body weight of 500 to 600 g, approximately double the body weights of their normal counterparts. The back (scruff of the neck) was used as the subcutaneous injection site in this study, in which the fat rats had significantly more subcutaneous fat, as evidenced by the skinfold thickness. To determine bioavailability (F), 36 μg/kg either PEG30-EPO or PEG40-EPO was administered intravenously and subcutaneously in the fat rats (n = 3/intravenous group; n = 6/subcutaneous group). As a control, the F of PEG40-EPO was also determined in the regular rats after 36 μg/kg intravenous and subcutaneous administration (n = 6/group). Blood samples were obtained from the jugular vein at specific time points. Serum samples were prepared and stored at −70°C until analysis.
PK Study in Beagle Dogs to Assess Impact of Physical Activity on Subcutaneous Absorption.
Beagle dogs with apparent different physical activity levels were used in the study. The dog's physical activity levels were divided into two categories based on cage side observation: high = excited barking, frequent jumping up and down, and vigorous tail wagging; low = quiet, usually laid down with occasional tail wagging.
To evaluate the impact of physical activity on subcutaneous absorption, 125I-labeled PEG30-EPO or PEG40-EPO (10 μg/kg, ∼10 μCi/kg) was administered subcutaneously to the posterior dorsal region, ∼10 cm in front of the tail. A modified hand-held sodium iodide detector was used to monitor disappearance of radioactivity from the injection site with minimal interference of signals from internal organs such as the thyroid. The sodium iodide detector was collimated with lead to limit the detection area to the injection site. Upon administration, the injection site was monitored at specific time points, and radiation measurements were recorded. Blood samples were obtained from the jugular vein at specific time points. Serum samples were prepared and stored at −70°C until analysis.
Analytical Method and PK Analysis.
When 125I-labeled compounds were used for animal studies, the total radioactivity levels in lymph, serum, urine, or feces samples were first determined by gamma counting (pre-TCA). The protein-associated radioactivity was subsequently quantified by TCA precipitation (Bensadoun and Weinstein, 1976). The TCA precipitation was conducted as described previously (Vugmeyster et al., 2010). Serum or lymph protein drug levels were calculated from TCA-precipitable (post-TCA) radioactivity using the specific activity of the dosing solutions with correction for 125I decay (nanogram-equivalents per milliliter). The percentage of TCA in a sample, calculated as post-TCA/pre-TCA, is an indication of integrity of the labeled protein, i.e., what percentage of the radioactivity is associated with protein, instead of being free iodine or very small degraded fragments.
When unlabeled compounds were used for animal studies, the serum drug levels were determined by an immunoassay based on the Quantikine IVD human erythropoietin kit and validated for quantifying PEG30-EPO and PEG40-EPO. The assay range was established with spiked quality control samples. The accuracy and precision were within 100 ± 20%, and the lower limit of quantitation was 0.1 ng/ml. The drug levels of selected 125I-labeled PEG30-EPO and PEG40-EPO serum/lymph samples were also determined by the immunoassay as a comparison to those calculated from post-TCA radioactivity. The results from both assays generally agree with each other, and the differences were less than 40%.
The serum drug concentrations were calculated for noncompartmental PK analysis using WinNonlin (Enterprise Version 5.2.1; Pharsight, Mountain View, CA). Both PEG30-EPO and PEG40-EPO exhibited approximately linear PK over the dose ranges (>10 μg/kg) used in our studies, and the systemic bioavailability (F, percentage) was calculated as follows: AUCsc · dosei.v./AUCi.v. · doses.c..
In Vitro Stability in Rat Subcutaneous Tissue Homogenate and Lymph Node Cell Suspensions.
The subcutaneous tissue from the back area of naive SD rats was excised and isolated with a scalpel. After mincing with a scissors, the subcutaneous tissue was mixed with 1.5× volumes of tissue weight of ice-cold homogenization buffer (50 mM Tris-HCl at pH7.4, 150 mM NaCl, 0.25 M sucrose, 1% Tween 20, and 1% Triton X-100) and homogenized with a Polytron homogenizer (Kinematica Inc., Newark, NJ) on ice. The lysate was centrifuged at 14,000g at 4°C for 20 min. The supernatant was removed for further analysis.
For lymph node cell suspensions, multiple lymph nodes from naive SD rats were excised. The cells were released by gently teasing using tweezers and then collected by straining through 40-μm cell strainers (BD Falcon, Franklin Lakes, NJ). The number of viable cells was determined by trypan blue dye exclusion with a hemacytometer (Gibco Cell Culture; Invitrogen, Carlsbad, CA). The specified numbers of viable cells were incubated with 125I-labeled PEG30-EPO or PEG40-EPO in a six-well tissue culture plate in RPMI 1640 medium with 10% fetal calf serum and 5% CO2 at 37°C for 24 h.
For in vitro stability experiments, 125I-labeled compounds were spiked into freshly prepared subcutaneous tissue homogenate, serum, lymph fluid, or lymph node cell suspensions and incubated at 37°C for 24 h. After incubation, SDS-polyacrylamide gel electrophoresis was performed to evaluate potential degradants, and the bands were detected by phosphorimaging (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
Results
Role of Lymphatic System in Subcutaneous Absorption of PEG30-EPO and PEG40-EPO in Rats.
To determine the contribution of the lymphatic system in subcutaneous absorption of macromolecules in commonly used preclinical species, we first used a rat thoracic LDC model established in-house. This model allows continuous thoracic duct lymph fluid collection in conscious and unrestrained SD rats for up to 7 days, considerably longer than what had been reported previously (Kagan et al., 2007; Kaminskas et al., 2009).
Before performing experiments in LDC rats, we examined the PK profiles of 125I-labeled PEG30-EPO and PEG40-EPO, for which the drug levels were calculated from post-TCA radioactivity, and found them to be similar to the corresponding PK profiles of unlabeled compounds, for which the drug levels were determined by a product-specific immunoassay: for PEG30-EPO, maximum serum concentration (Cmax) was 114 ± 30 versus 73 ± 26 ng/ml and AUC0–168 h was 7649 ± 1566 versus 7059 ± 3325 h · ng/ml after 25 μg/kg s.c. dosing in rats; for PEG40-EPO, Cmax was 85 ± 42 versus 84 ± 26 ng/ml and AUC0–168 h was 5013 ± 3635 versus 5193 ± 1493 h · ng/ml after 36 μg/kg s.c. dosing in rats. These results suggested that 125I-labeling did not significantly change the PK properties of PEG30-EPO and PEG40-EPO and that the drug levels calculated from post-TCA radioactivity can generally be considered to be representative of those determined by immunoassay. We also determined the systemic bioavailability of PEG30-EPO and PEG40-EPO after subcutaneous administration using unlabeled compounds in intact control SD rats, and they were 38 and 30%, respectively (Table 1). In addition, our PK studies with unlabeled compounds had demonstrated that the clearance of PEG40-EPO is linear between 4 and 36 μg/kg in rats (2.0–2.2 ml/kg per hour). The clearance of PEG30-EPO (CERA) was also reported to be approximately linear between 2.5 and 25 μg/kg in rats and 3 and 7.5 μg/kg in dogs (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000739/WC500033669.pdf).
PK parameters of PEG30-EPO and PEG40-EPO in the thoracic LDC and control SD rats after subcutaneous administration
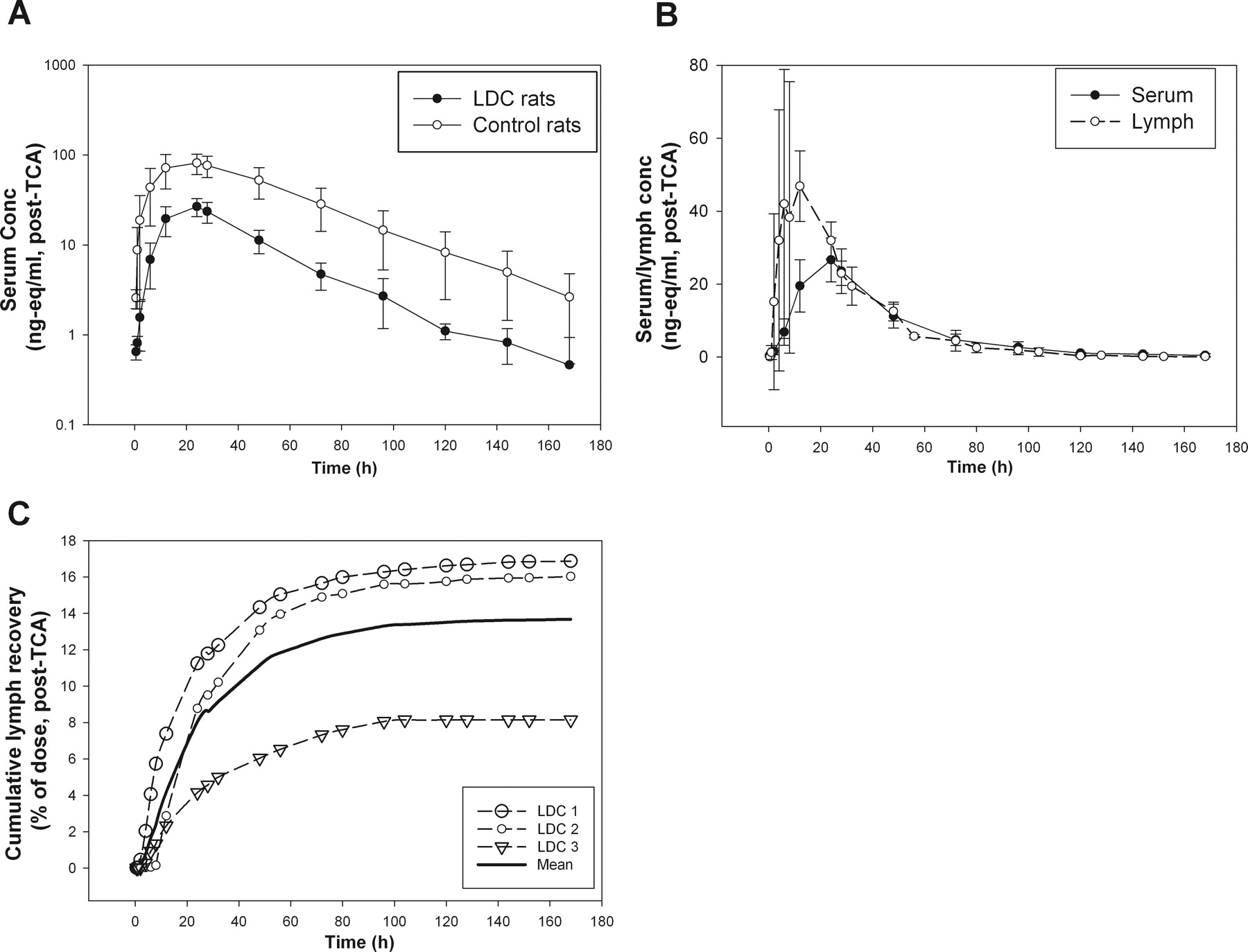
The choice of subcutaneous injection site is very important for studying lymphatic contribution in rat thoracic LDC models. For all LDC rat studies described, we used the lower hind leg (just below the knee) as the subcutaneous injection site to evaluate the contribution of the lymphatic system because this is the only subcutaneous site at which most of the lymph fluid would drain through the thoracic LDC site (Tilney, 1971). 125I-labeled PEG30-EPO was dosed subcutaneously in the lateral left lower hind leg in the thoracic LDC rats (n = 6) and in the sham-operated control rats (n = 3). PEG30-EPO levels (nanogram equivalents per milliliter) in the serum were calculated from post-TCA radioactivity levels, and the serum concentration-time profiles are shown in Fig. 1A. Both Cmax and serum exposure (AUC0–168 h) of PEG30-EPO were significantly reduced (∼70%) in the thoracic LDC rats compared with those in the sham-operated control rats (Fig. 1A). The serum and lymph concentration-time profiles from the thoracic LDC rats are compared in Fig. 1B. During the absorption or ascending phase of serum concentration profiles (<24 h), the drug levels were significantly higher in the lymph than that in the serum. Even though no intravenous study was conducted in LDC animals, the comparison between lymph and serum concentration-time profiles demonstrated that the drug exposure in lymph preceded the systemic exposure. This result clearly indicated that the observed lymph radioactivity at early time points (<24 h) mostly came from subcutaneous absorption not distribution.
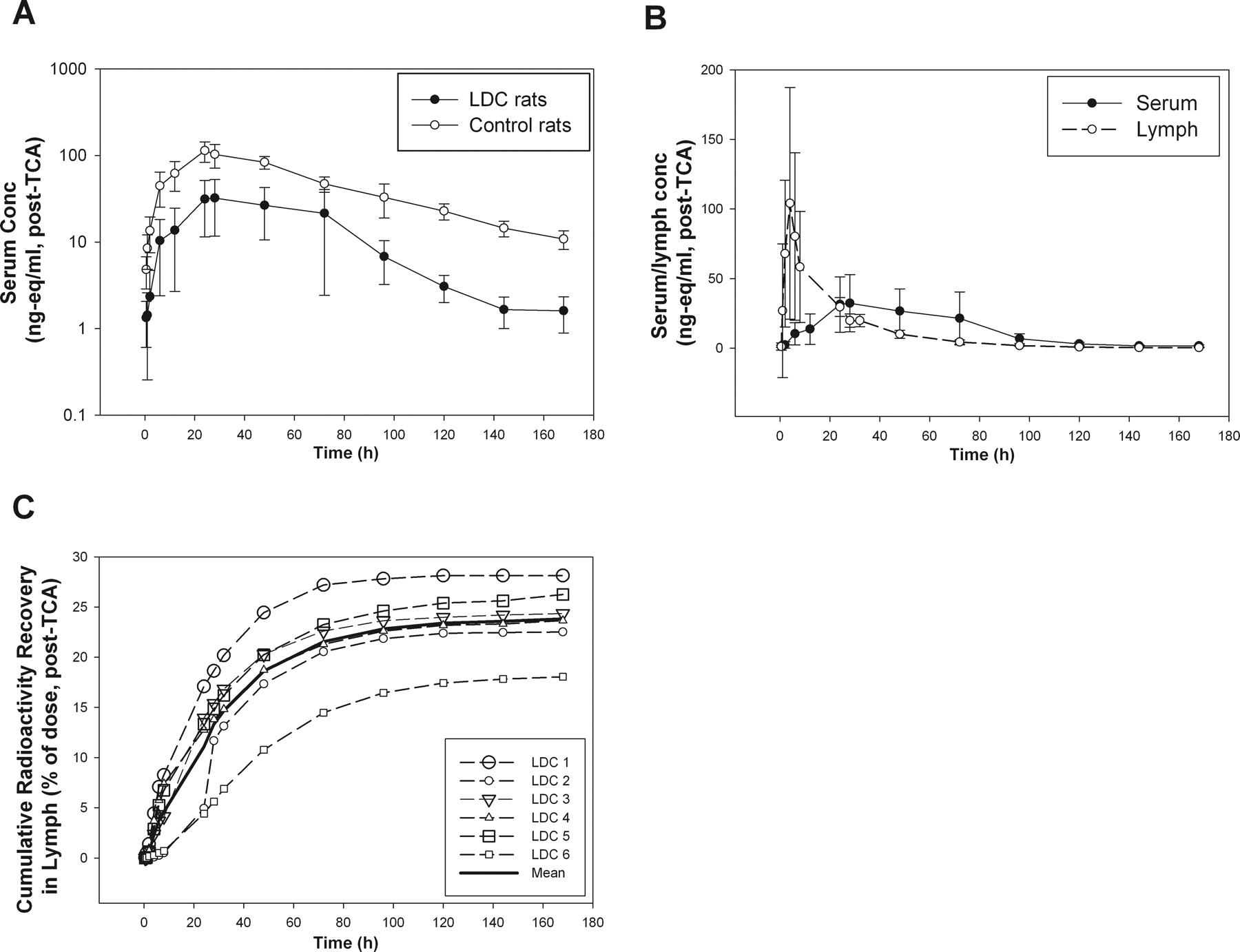
A, mean (SD) serum concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG30-EPO after subcutaneous administration in the thoracic LDC rats (n = 6) and sham-operated control rats (n = 3). B, mean (SD) serum and lymph concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG30-EPO in the thoracic LDC rats (n = 6). C, cumulative recovery of dosed radioactivity (post-TCA) in lymph collected from the thoracic LDC rats (mean and individual data).
We also collected radioactivity in the lymph over the 7-day study period, and the results are summarized in Table 1. When the total radioactivity (pre-TCA) was examined, approximately 31% (31.1 ± 3.7%) of the dose was recovered in the thoracic duct lymph. When the post-TCA radioactivity (protein-associated) was counted, approximately 24% (23.8 ± 3.5%) of the dose was recovered. The profiles of post-TCA cumulative lymph recovery in individual animals are also shown in Fig. 1C. On the basis of the post-TCA lymphatic recovery, the lymph contribution was estimated to be ∼60 to 70% of what was available systemically in intact animals (F of ∼38%). This estimate is consistent with the observed ∼70% reduction of serum exposure and consequently the reduced F (to 10%) of PEG30-EPO in the thoracic LDC rats.
As shown in Table 1, nearly 80% of dosed radioactivity was recovered, primarily in urine and minimally in feces, in intact control animals. This result indicates that at least 80% of this protein drug was absorbed from the injection site. The total radioactivity recovery (lymph + excreta) in the LDC animals was comparably high (∼72%), with nearly 40% of the dose recovered in urine and ∼30% of dose in the lymph (Table 1). Considering that the total radioactivity recoveries were significantly higher than the observed F of ∼38% in intact animals, these results suggest potential catabolism of PEG30-EPO after subcutaneous administration before reaching the systemic circulation.
125I-labeled PEG40-EPO, a PEGylated EPO produced in-house, was also examined in the rat thoracic LDC model (n = 3). Similar to the observation with PEG30-EPO, ∼70% reduction in Cmax and serum exposure (AUC0–168 h) of PEG40-EPO in the thoracic LDC rats was observed (Fig. 2A). Likewise, the post-TCA radioactivity levels were also significantly higher in the lymph than in the serum during the absorption phase (<24 h), supporting the fact that the observed lymph radioactivity at early time points came from subcutaneous absorption not distribution (Fig. 2B). In addition, a significant amount of dosed radioactivity (approximately 20% as pre-TCA and up to 17% as post-TCA) was recovered in the lymph over the period studied, which represents ∼60% of the F value of ∼30% (Table 1; Fig. 2C).

A, mean (SD) serum concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG40-EPO after SC administration in the thoracic LDC rats (n = 3) and sham-operated control rats (n = 3). B, mean (SD) serum and lymph concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG40-EPO in the thoracic LDC rats (n = 3). C, cumulative recovery of dosed radioactivity (post-TCA) in lymph collected from the thoracic LDC rats (mean and individual data).
Catabolism after Subcutaneous Administration in Rats.
We next examined potential catabolic activities after subcutaneous administration as possible causes for the significantly lower than expected F, on the basis of the total radioactivity recovered in rats. We first assessed potential catabolic activity at the subcutaneous injection site using freshly prepared rat subcutaneous tissue homogenate from naive animals. As shown in Fig. 3A, distinctive degradation products were observed for both PEG30-EPO and PEG40-EPO after the 24-h incubation, suggesting that there was potential catabolism of these molecules at the subcutaneous injection site. The observed catabolic activity appeared to be specific to the subcutaneous tissue, because similar experiments with freshly prepared rat serum or lymph did not reveal any catabolic activity when either was incubated with PEG30-EPO and PEG40-EPO (data not shown). PEG40-EPO appeared to have slightly more breakdown fragments than PEG30-EPO, but no apparent degradation was seen for the vast majority (90–95%) of both compounds over the 24-h incubation period (Fig. 3A). Multiple subcutaneous tissue homogenate preparation methods had been evaluated, and the one used here exhibited the highest catabolic activity. Despite our efforts to avoid it, potential loss of catabolic activity during the subcutaneous tissue homogenization and/or incubation processes could not be ruled out.
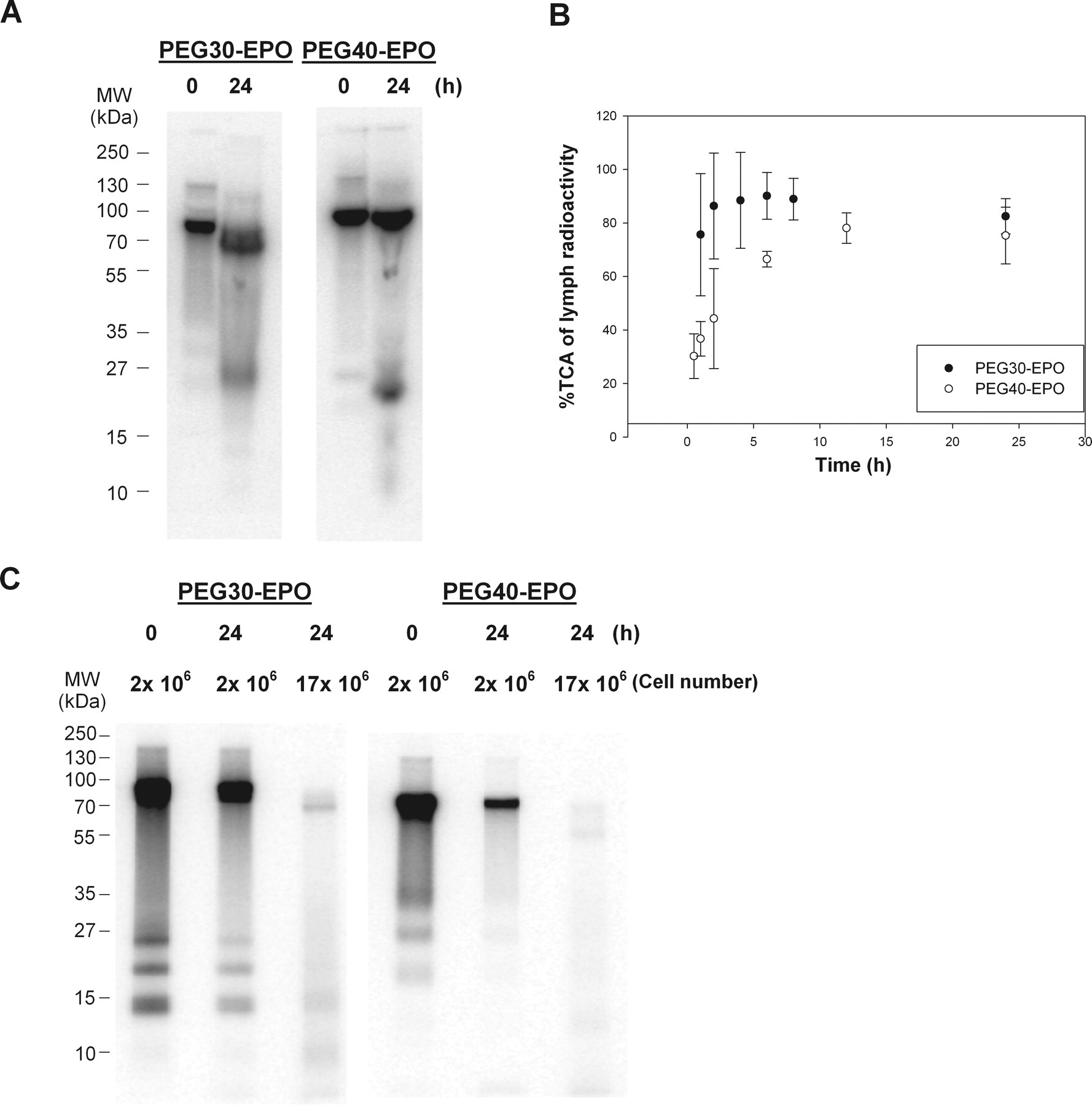
A, in vitro stability of PEG30-EPO and PEG40-EPO in rat SC tissue homogenates for 24 h. B, percentage of TCA of dose-related radioactivity in lymph collected from the thoracic LDC rats. C, in vitro stability of PEG30-EPO and PEG40-EPO in rat lymph node cell suspensions.
We also investigated potential catabolism during the lymphatic transport as another possible mechanism for the relatively low F in rats. Charman et al. (2000) had presented evidence of catabolism during lymphatic transport for hGH, but it has remained the only such example. In this study, we first examined the lymph collected from the thoracic LDC rats after subcutaneous administration. Of interest, considerable amounts of non-TCA precipitable, dose-related radioactivity were found, especially during early time points (i.e., <8 h) (Fig. 3B). The presence of non-TCA-precipitable radioactivity in the lymph suggested potential catabolism during lymphatic transport, because these non-TCA precipitable, small fragments were not expected to be absorbed via the lymphatic system if they were generated at the subcutaneous injection site. It was also interesting to note that the percentage of TCA for PEG30-EPO was considerably higher than that for PEG40-EPO at early time points (<8 h, p < 0.01) (Fig. 3B), which suggested that the extent of catabolism during lymphatic transport could be compound-dependent.
Consistent with the previous report for hGH (Charman et al., 2000), we did not find any catabolic activity when freshly prepared rat lymph was incubated with PEG30-EPO and PEG40-EPO (data not shown). To investigate the potential source of catabolism during lymphatic transport, we prepared lymph node cell suspensions as described under Materials and Methods. Lymph node cell suspensions instead of lymph node homogenate were used because we anticipated that the catabolic activity probably came from live phagocytic cells residing in the lymph nodes. As shown in Fig. 3C, profound catabolic activity was observed when lymph node cell suspensions were incubated with 125I-labeled PEG30-EPO and PEG40-EPO under cell culture conditions: the amounts of protein corresponding to the original product were significantly reduced after the 24-h incubation with 2 × 106 live cells, and there was almost none left after the incubation with 17 × 106 live cells. Again, PEG40-EPO appeared to disappear slightly faster than PEG30-EPO (Fig. 3C). No distinctive degradant was observed after the incubation with lymph node cell suspensions, which was different from that for the subcutaneous tissue homogenates.
Subcutaneous Absorption in Fat Rats.
We next examined whether subcutaneous absorption in fat rats was similar to that in regular rats, because adipose tissue had been associated with poor lymphatic drainage (Ryan, 1995; Swartz, 2001), and body weight is a common negative covariate for F after subcutaneous administration in humans (Macdougall et al., 1991; Silva et al., 2006; Olsson-Gisleskog et al., 2007).
A comparison of the PK profiles of PEG40-EPO in fat rats and regular rats after intravenous and subcutaneous administration is shown in Fig. 4. Because the fat rats had ∼2-fold higher body weights than their normal counterparts, they received approximately two times more of the drug when a body weight-normalized dose (36 μg/kg) was given. Consistent with the finding that obese rats have a comparable blood volume and lower blood volume-to-body weight ratio than regular rats (Schreihofer et al., 2005), the serum exposure of PEG40-EPO after intravenous administration was ∼2-fold higher in fat rats than that in regular rats (Fig. 4). However, PEG40-EPO exhibited ∼2-fold lower exposure in the fat rats after subcutaneous administration, despite the fact that ∼2-fold more of the drug was administered (Fig. 4). Overall, PEG40-EPO exhibited significantly lower F in fat rats compared with that in regular rats (6 versus 25%).

Mean (SD) serum concentration (immunoassay, nanograms per milliliter) versus time profiles of PEG40-EPO after intravenous (n = 3) and SC (n = 6) administration in regular and fat rats.
The F of PEG30-EPO was also determined in fat rats after intravenous and subcutaneous administration, and the F was found to be ∼19% (data not shown), which was significantly lower than the reported F of 31 to 45% in regular rats (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000739/WC500033669.pdf), although the difference between fat rats and regular rats appeared to be less for PEG30-EPO (∼2-fold) than that for PEG40-EPO (∼4-fold).
Subcutaneous Absorption of PEG40-EPO in Dogs.
We also conducted limited studies in beagle dogs to determine whether the subcutaneous absorption mechanism is conserved across species. First, 125I-labeled PEG40-EPO was used to study the contribution of the lymphatic system in subcutaneous absorption using a thoracic LDC model in beagle dogs. The results obtained in LDC dogs were similar to those obtained in LDC rats, despite the limited number of animals being used (one after subcutaneous administration and one after intravenous administration) (Fig. 5).
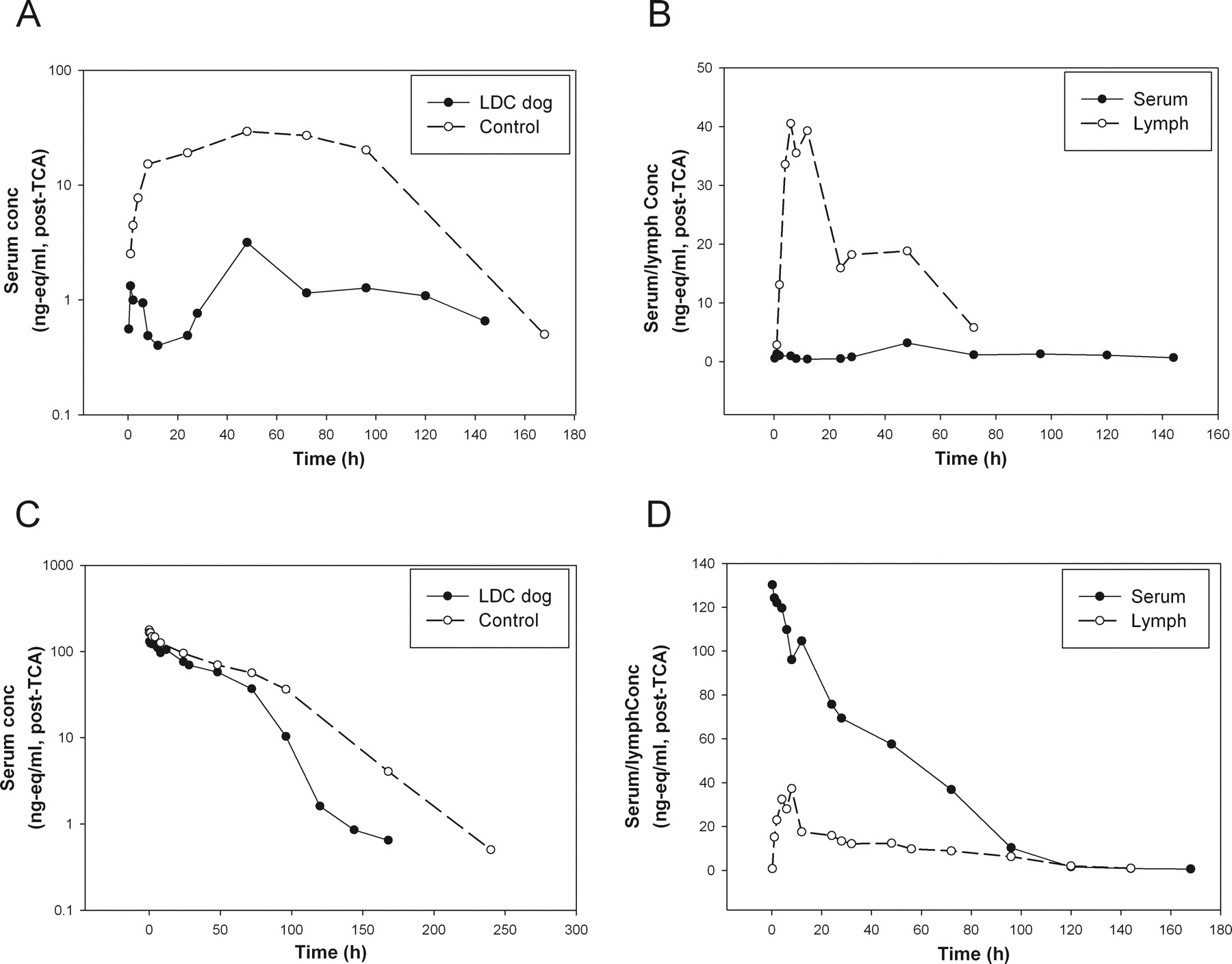
A, serum concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG40-EPO after subcutaneous administration in the thoracic LDC (n = 1) and control (n = 3, mean) dogs. B, serum and lymph concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles in the thoracic LDC dog after subcutaneous administration. C, serum concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles of PEG40-EPO after intravenous administration in the thoracic LDC (n = 1) and control (n = 3, mean) dogs. D, serum and lymph concentrations (nanogram equivalents per milliliter, post-TCA) versus time profiles in the thoracic LDC dog after intravenous administration.
After subcutaneous administration, the serum exposure and Cmax of PEG40-EPO were reduced significantly in the thoracic LDC dog compared with those in the control dogs (Fig. 5A). In addition, the post-TCA radioactivity levels in the lymph were significantly higher than those in serum, especially during the absorption phase (Fig. 5B). Overall, 20% of the dosed radioactivity (post-TCA) was recovered in the thoracic duct lymph over the 7-day study period after subcutaneous administration.
In the thoracic LDC dog that received an intravenous dose, the serum PK profile only appeared to deviate significantly from that of the control animals after 72 h (Fig. 5C). In contrast to all the subcutaneous studies in LDC animals, the post-TCA radioactivity levels in lymph were considerably lower than those in serum at all early time points (Fig. 5D), consistent with the expectation that the lymph radioactivity came from distribution after intravenous administration. In this LDC dog, much lower radioactivity recovery (∼6% after intravenous versus 20% after subcutaneous) was seen in the lymph, presumably through distribution. Although it was difficult to quantify the relative difference in the lymph recovery after the intravenous versus subcutaneous doses because we examined only one LDC dog in each arm, the lymph recovery results nevertheless are in line with the differences observed in the plasma profiles between the two administration routes and suggested that the lymphatic system played an important role in the subcutaneous absorption, not just solely in redistribution, of this macromolecule in dogs.
We also examined the impact of physical activity, another physiologic factor pertinent to lymphatic transport, on subcutaneous absorption in dogs, because physical activity is known to have a direct impact on the lymph flow rate (Swartz, 2001; Downey et al., 2008) and it is possible to identify dogs with apparent differences in physical activities (see Materials and Methods). Five dogs with different (high or low) physical activity levels were used in the study. They were dosed subcutaneously with 125I-labeled PEG30-EPO (1× high, 2× low) or PEG40-EPO (1× high, 1× low) ∼10 cm in front of the tails. This subcutaneous injection site was selected to facilitate monitoring of the disappearance of radioactivity from the injection site with minimal interference of signals from internal organs such as the thyroid. As shown in Fig. 6, A and B, dogs with higher levels of physical activity exhibited a faster disappearance of radioactivity from the injection site for both compounds and correspondingly significantly faster appearance of radioactivity in the systemic circulation and higher Cmax and serum exposure than the dogs with lower physical activity levels (Fig. 6, C and D). Even though we have yet to establish an objective method to quantify the physical activity levels, these results clearly demonstrated the impact of physical activity on subcutaneous absorption in this species.

Disappearance of dosed radioactivity from subcutaneous injection sites in dogs with different physical activity levels after subcutaneous administration (10 μg/kg, ∼10 μCi/kg): A, PEG30-EPO; B, PEG40-EPO. Serum concentrations (post-TCA, nanogram equivalents per milliliter) versus time profiles: C, PEG30-EPO; D, PEG40-EPO. The dog physical activity levels (high or low) were as labeled in the graph.
Discussion
Using the thoracic LDC models established in-house and PEGylated EPOs as model compounds, we showed that these macromolecules were mostly absorbed via the lymphatic system after subcutaneous administration in rats and dogs. The thoracic LDC animals exhibited ∼70% lower serum exposure than the control animals and up to 70% of the systemically available drugs was recovered in the lymph. Moreover, after subcutaneous administration, lymph exposure was found to precede systemic exposure in lymph duct-cannulated animals, indicating that the radioactivity in the lymph during the ascending phase mostly came from absorption not distribution. These results agreed well with previous reports in the sheep lymph duct cannulation model (Supersaxo et al., 1990; Porter and Charman, 2000; Porter et al., 2001). Taken together, our data and previous reports suggest that the role of the lymphatic system in subcutaneous absorption is probably conserved in these preclinical species.
However, our results contradicted a few previous reports, which suggested minimal lymphatic contribution to subcutaneous absorption in rats and rabbits (Bocci et al., 1986, 1988; Kojima et al., 1988; Kagan et al., 2007). Although factors such as the success of lymph duct cannulation models or the selection of model compounds, e.g., PEGylated versus non-PEGylated proteins, could have contributed to the observed differences, one likely explanation for the discrepancy is the choice of subcutaneous injection sites. In smaller-sized laboratory animals, the selection of the subcutaneous injection site is important for studying lymphatic contribution. Given the size of a rat, technically it is only feasible to cannulate the thoracic duct at a site just above the cisterna chyli (Ionac, 2003; Kagan et al., 2007; Kaminskas et al., 2009). The lymphatic drainage routes in adult laboratory rats were mapped in detail by Tilney (1971). In contrast to the conventional wisdom, most of the commonly used subcutaneous injection sites, including the sites used in those previous reports (thigh), drained via the brachial, inguinal, or axillary lymph nodes, which eventually enter the systemic circulation through the subclavian duct, bypassing the thoracic LDC site. The only subcutaneous injection site that would drain mostly through the thoracic LDC site is the lower hind leg region (Tilney, 1971).
Using the lower hind leg area as a subcutaneous injection site, we found that the cumulative recovery of dose in the thoracic duct lymph was much higher than what had been reported previously, up to 28% of the dose or up to 70% of the systemically available dose. Even when the lower hind leg region was used as the subcutaneous injection site, a fraction of the dose was still expected to travel down the inguinal lymph node-subclavian duct path (Tilney, 1971). Therefore, the ∼10% of drugs remaining systemically available in the thoracic LDC rats (Figs. 1A and 2A) did not necessarily enter the systemic circulation via blood capillaries but could also have been absorbed via a lymphatic route that bypassed the thoracic LDC site.
Of interest, the only previous work that reported recovery of a significant percentage of subcutaneous dosed radioactivity in the thoracic duct lymph of rats also used the lower hind leg area as the subcutaneous injection site (Kaminskas et al., 2009). In larger animals such as sheep, it was possible to recover a significant amount of the subcutaneous dose in the thoracic duct lymph even when injection sites other than lower hind legs were used (Kota et al., 2007). One possible reason is that in sheep, one can cannulate the thoracic duct at a position very close to where it enters the systemic circulation (Kota et al., 2007). It could also be related to interspecies differences in lymphatic drainage pathways.
In this report, we present direct evidence for near-complete absorption of two PEGylated proteins from the injection site after subcutaneous administration and the catabolism at the subcutaneous injection site and during lymphatic transport as a likely cause for the limited subcutaneous bioavailability. The loss of bioavailability after subcutaneous administration had mostly been empirically attributed to loss/degradation at the injection site (Lee, 1988; Mrsny and Daugherty, 2009). Using rat subcutaneous tissue homogenates, we confirmed that there indeed was catabolic activity in the subcutaneous tissue, at least for the model compounds we examined. In addition, a previous report had demonstrated significant loss of bioavailability during lymphatic transport of hGH in the sheep lymph duct cannulation model, in which 61.7% of drug was recovered in the peripheral lymph, whereas only 8.6% was recovered in the central lymph (Charman et al., 2000). Here, we observed a considerable amount of non-TCA precipitable small fragments in the thoracic duct lymph after subcutaneous administration, which indicated catabolism during lymphatic transport, because small fragments generated at the subcutaneous site are expected to be taken up by the blood capillary directly (Porter and Charman, 2000). In our study, we also found high catabolic activity with lymph node cell suspensions, suggesting that the lymph node is a potential site for loss of bioavailability during lymphatic transport. Lymph nodes are part of our immune system, designed to filter and trap foreign particles (Gonzalez et al., 2010). Although our finding is not entirely surprising, to the best of our knowledge, this is the first demonstration of such a phenomenon. Of interest, we encountered high variability in the catabolic activity for our lymph node cell suspension preparations (data not shown). It remains to be determined whether this was due to the technical variability or intrinsic variability of the catabolic activities of lymph nodes.
The differences we observed between PEG30-EPO and PEG40-EPO are interesting, because they suggested that relatively minor differences in molecular characteristics such as glycosylation or the size of PEG, may have an impact on subcutaneous absorption/stability. However, the differences observed here are relatively small and we have no reason to believe that they would be clinically important.
Many physiologic factors such as age, body weight, and injection sites have been reported as covariates for subcutaneous bioavailability in humans (Macdougall et al., 1991; Chan et al., 2003; Silva et al., 2006; Fishbane et al., 2007; Olsson-Gisleskog et al., 2007; Kakkar et al., 2011). In this investigation, we show that physiologic factors such as fat content and physical activity can have a profound impact on subcutaneous absorption of PEGylated proteins in preclinical species. The impacts we observed were consistent with their expected impact on the lymphatic transport; i.e., factors associated with faster lymphatic transport are associated with faster absorption and higher bioavailability and vice versa. In addition to the results presented here, we also found that factors such as dose, dosing volume/dosing concentration, and injection site, all could affect subcutaneous absorption of a macromolecule (data not shown). For all the covariates of subcutaneous absorption we evaluated, a common theme is that faster absorption usually leads to higher bioavailability. A recent study on subcutaneous absorption of rituximab in rats reported similar findings (Kagan et al., 2012). Although a slower absorption itself should not be the reason for lower bioavailability, it is conceivable that the rate of transport from the injection site and then through the lymphatic system determines the extent of catabolism before systemic exposure. For the purpose of comparing bioavailability, these results illustrated the importance of controlling these potential covariants of subcutaneous absorption in preclinical and/or clinical studies, because the magnitude of their impact can be significant. Given our observations, we should also actively seek preclinical models more reflective of the intended clinical settings to better assess the impact of a particular physiologic factor for the compound of interest.
Human PK of biologic drugs after intravenous administration can usually be predicted with reasonable confidence using allometric scaling (Mordenti et al., 1991; Mahmood, 2004; Wang and Prueksaritanont, 2010), but it is much more challenging to predict their subcutaneous absorption process in humans. There are fundamental differences in subcutaneous tissue structure between preclinical species and humans (Magnusson et al., 2001). Therefore, prediction of human PK after subcutaneous administration probably will rely on mechanistic understanding of the subcutaneous absorption process rather than on empirical scaling methods. For example, in vitro tools such as the subcutaneous tissue homogenate and lymph node cell suspensions should be very useful in assessing catabolic activity that could play a major role in determining bioavailability of a given macromolecule compound. These systems cannot necessarily be used to quantitatively assess bioavailability but rather can be used for comparison purposes and rank-ordering of compounds. They are also one way to address potential interspecies differences in bioavailability associated with interspecies difference in catabolic stability. Of interest, our results suggest that the impact of physiologic factors on subcutaneous absorption can be compound-dependent; i.e., the intrinsic “stability” of a molecule can affect its sensitivity to such physiologic features. We believe that an appropriate use of preclinical models and in vitro tools should facilitate mechanistic understanding and aid human PK prediction of bioavailability and potential variability. In combination with conventional PK studies in preclinical species, the tools presented here can provide useful information for candidate selection, human PK prediction, clinical study design, and risk mitigation for given biologic drug candidates.
Authorship Contributions
Participated in research design: W. Wang, Chen, Liu, Hamuro, and Prueksaritanont.
Conducted experiments: Chen, Shen, Cunningham, Fauty, Michel, B. Wang, Hong, Adreani, Nunes, Johnson, and Zou.
Contributed new reagents or analytic tools: Chen, Yin, and Groff.
Performed data analysis: W. Wang and Chen.
Wrote or contributed to the writing of the manuscript: W. Wang and Prueksaritanont.
Acknowledgments
We thank Lorraine Lipfert for excellent technical assistance in the preparation of 125I-labeled compounds, Carmen Fernandez-Metzler for reviewing the manuscript, Marissa Vavrek for coordination of in vivo studies, and Huijuan Li for providing PEG40-EPO.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- PK
- pharmacokinetics
- LDC
- lymph duct cannulation
- EPO
- erythropoietin
- PEG
- polyethylene glycol
- CERA
- continuous erythropoietin receptor activator
- SD
- Sprague-Dawley
- TCA
- trichloroacetic acid
- AUC
- area under the serum concentration-time curve
- hGH
- human growth hormone.
- Received November 7, 2011.
- Accepted February 10, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}