


Abstract
Breast cancer resistance protein (BCRP; ABCG2) limits intestinal absorption of low-permeability substrate drugs and mediates biliary excretion of drugs and metabolites. Based on clinical evidence of BCRP-mediated drug-drug interactions (DDIs) and the c.421C>A functional polymorphism affecting drug efficacy and safety, both the US Food and Drug Administration and European Medicines Agency recommend preclinical evaluation and, when appropriate, clinical assessment of BCRP-mediated DDIs. Although many BCRP substrates and inhibitors have been identified in vitro, clinical translation has been confounded by overlap with other transporters and metabolic enzymes. Regulatory recommendations for BCRP-mediated clinical DDI studies are challenging, as consensus is lacking on the choice of the most robust and specific human BCRP substrates and inhibitors and optimal study design. This review proposes a path forward based on a comprehensive analysis of available data. Oral sulfasalazine (1000 mg, immediate-release tablet) is the best available clinical substrate for intestinal BCRP, oral rosuvastatin (20 mg) for both intestinal and hepatic BCRP, and intravenous rosuvastatin (4 mg) for hepatic BCRP. Oral curcumin (2000 mg) and lapatinib (250 mg) are the best available clinical BCRP inhibitors. To interrogate the worst-case clinical BCRP DDI scenario, study subjects harboring the BCRP c.421C/C reference genotype are recommended. In addition, if sulfasalazine is selected as the substrate, subjects having the rapid acetylator phenotype are recommended. In the case of rosuvastatin, subjects with the organic anion–transporting polypeptide 1B1 c.521T/T genotype are recommended, together with monitoring of rosuvastatin's cholesterol-lowering effect at baseline and DDI phase. A proof-of-concept clinical study is being planned by a collaborative consortium to evaluate the proposed BCRP DDI study design.
Introduction
Breast cancer resistance protein (BCRP; ABCG2) is an ATP-binding cassette efflux transporter comprising a homodimer of two half subunits (Giacomini et al., 2010). BCRP is expressed in the small intestine, liver, blood-brain barrier, testis, placenta, and mammary glands (Endres et al., 2006) and limits systemic and organ exposure of relevant substrates. BCRP transports endogenous and exogenous substrates with diverse physicochemical properties, which generally include a positive and/or negative charge but otherwise span a large range of molecular weight, lipophilicity, and permeability (Mao and Unadkat, 2005; Ni et al., 2010; Robey et al., 2011). However, the impact of BCRP on pharmacokinetics (PK) is the most pronounced for compounds with low passive permeability (Poirier et al., 2014).
From a drug disposition perspective, BCRP functions primarily as an apical efflux pump in enterocytes, attenuating intestinal absorption of low-permeability substrate drugs, and as a canalicular efflux pump, transporting substrates from hepatocytes into bile. Unlike intestinal BCRP, which is rate-determining in the absorption of low-permeability substrate drugs, in the liver, uptake is generally rate-determining in hepatic clearance of drugs that predominantly rely on carrier-facilitated transport into the liver prior to excretion into bile via BCRP. However, when a basolateral efflux mechanism is operational [e.g., multidrug resistance–associated protein (MRP) 4], BCRP biliary excretion can become rate-determining in systemic drug clearance when it is impaired to a sufficiently large extent (Pfeifer et al., 2013a,b). Such shifting of the rate-determining step in hepatobiliary clearance is a kinetically complex phenomenon and is best described with both systemic PK data as well as a measure of hepatic drug exposure, which in humans can be assessed directly (e.g., imaging of a metabolically stable drug) or indirectly using a hepatic pharmacodynamic (PD) biomarker (e.g., cholesterol for statins).
Pharmacogenetic studies identified a c.421C>A (p.Q141, rs2231142) single nucleotide polymorphism (SNP) of BCRP that results in reduced activity (Giacomini et al., 2013). In vitro studies demonstrated that this BCRP variant maintains mRNA expression, but 50%–70% reduced protein expression and function was observed due to enhanced susceptibility to proteasomal degradation (Kondo et al., 2004; Furukawa et al., 2009). Several marketed drugs, including sulfasalazine, rosuvastatin, and atorvastatin, have increased systemic exposure due to impaired BCRP activity in carriers of this functional polymorphism (Giacomini et al., 2013). For example, rosuvastatin demonstrated 2.4-fold-higher exposure in homozygous BCRP c.421A/A individuals compared with homozygous c.421C/C individuals (Keskitalo et al., 2009b). In addition to changes in the PK profile, clinical studies involving BCRP c.421A/A homozygous carriers demonstrated an increased incidence of gefitinib side effects (Cusatis et al., 2006).
BCRP has been demonstrated to be an important mediator of drug-drug interactions (DDIs) in humans (Giacomini et al., 2010). For example, the herbal product curcumin increased sulfasalazine oral exposure 3.2-fold. This clinical DDI has been attributed to BCRP inhibition, because it could be replicated in wild-type but not Bcrp-knockout mice, clearly proving Bcrp to be the DDI mechanism (Shukla et al., 2009; Kusuhara et al., 2012).
The clinical evidence of BCRP inhibition as a mechanism of DDIs with potential safety and efficacy consequences prompted the International Transporter Consortium to highlight BCRP as an important transporter to evaluate during drug development (Giacomini et al., 2010). Subsequently, the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) recommended that all investigational drugs should be evaluated as substrates (victims) or inhibitors (perpetrators) of BCRP and, when warranted, that victim and perpetrator DDI potential should be characterized in humans (US Food and Drug Administration, 2012; European Medicines Agency, 2013). The exception to the BCRP substrate evaluation is when the drug candidate meets biopharmaceutical classification system 1 criteria, where intestinal solubility and permeability are too high for oral absorption to be attenuated by BCRP efflux (US Food and Drug Administration, 2012; European Medicines Agency, 2013). These regulatory recommendations are reasonable and justified by overall clinical evidence supporting BCRP as a contributor to, although typically not the sole site of, victim and perpetrator DDIs.
Many drugs from various therapeutic areas have been identified as substrates or inhibitors of BCRP in vitro, yet clinical DDIs attributed directly and specifically to BCRP are limited due to overlap with other transport, as well as metabolic, pathways (Poirier et al., 2014). For example, the 8-fold increase in atorvastatin exposure during tipranavir and ritonavir coadministration was attributed mechanistically to inhibition of BCRP, organic anion–transporting polypeptide (OATP) 1B1, and CYP3A4 (Pham et al., 2009); however, quantification of the relative contribution of each of these pathways to the overall DDI is challenging. Because well characterized clinical BCRP substrate probes and inhibitors are lacking, DDIs attributed solely to BCRP are rare. As a consequence, a consensus is lacking on how to optimally evaluate clinical BCRP victim and perpetrator DDI potential.
Recognizing the challenges faced by the pharmaceutical industry regarding the clinical evaluation of BCRP-mediated drug disposition and DDIs, the intent of this review is 1) to identify the best available clinical probes and inhibitors that can be used to understand the contribution of BCRP to intestinal, hepatic, or overall systemic drug disposition in humans; and 2) to propose a clinical study design to test the validity of the selected substrate/inhibitor pairs to evaluate BCRP-mediated DDIs, as well as delineate organ-specific BCRP contributions in humans.
Clinical Relevance of BCRP Polymorphisms
Impaired BCRP activity is associated with a nonsynonymous SNP, c.421C>A (Furukawa et al., 2009). Clinically, this polymorphism is linked to increased exposure to rosuvastatin, sunitinib, an active metabolite of leflunomide, sulfasalazine, flavopiridol, diflomotecan, and gefitinib (Giacomini et al., 2013). For example, homozygous carriers of the variant allele (c.421A/A) showed up to a 3.5-fold increase in sulfasalazine oral exposure compared with homozygous carriers of the reference allele (c.421C/C) (Yamasaki et al., 2008; Kusuhara et al., 2012). Clinical examples of elevated statin exposure in homozygous carriers of the c.421C>A SNP range from 1.7-fold increase for both atorvastatin and fluvastatin to 2.4-fold increase for rosuvastatin (Keskitalo et al., 2009a,b).
In general, the effect of the BCRP c.421C>A variant on the PK of most BCRP substrates has been relatively modest; however, a major limitation of many of these studies is the low number of subjects homozygous for this SNP due to low population frequency of the recessive allele. For example, intravenous diflomotecan exhibited 3-fold higher intravenous exposure in c.421C/A (n = 5) versus c.421C/C carriers (n = 15) but no change in area under the concentration-time curve (AUC) following oral drug administration (Sparreboom et al., 2004), while rosuvastatin exposure was 1.8-fold higher in a combined cohort of c.421C/A and A/A (n = 5 C/A and n = 2 A/A) versus c.421C/C carriers (n = 7) (Zhang et al., 2006) (Table 1). In contrast, no effect of the c.421C/A SNP was observed on the oral PK of the following BCRP substrates: nitrofurantoin, lamivudine, pravastatin, pitavastatin, and intravenous irinotecan (Ferrante et al., 1991; de Jong et al., 2004; Han et al., 2007; Ieiri et al., 2007; Jada et al., 2007; Adkison et al., 2008; Keskitalo et al., 2009a; Zhou et al., 2013). Lack of effect of BCRP c.421C>A on pravastatin exposure (Keskitalo et al., 2009a) stands in contrast to rosuvastatin (which has comparable low passive permeability to pravastatin) and supports the hypothesis that P-glycoprotein (P-gp) and MRP2 play more prominent roles than BCRP in pravastatin absorption.
Summary of rosuvastatin BCRP pharmacogenomic studies and associated effect on its PK
The above examples highlight the critical importance of thoughtful design of BCRP pharmacogenetic studies when they are intended to be used in lieu of traditional DDI inhibitor studies to delineate the victim DDI potential of a BCRP substrate drug. As demonstrated by the above examples, the number of study subjects in typical DDI studies is not high enough to randomly enroll enough carriers of the BCRP c.421A/A and c.421C/A genotypes, making a post hoc comparison of BCRP genotype effects impossible. As such, when BCRP pharmacogenetic studies are intended to be used as a substitute for a DDI study with a BCRP inhibitor, subjects must be genotyped during enrollment, and the study should be powered with an equal and adequate number of subjects in each of the three groups (C/C, C/A, and A/A). The two sulfasalazine BCRP pharmacogenetic studies by Yamasaki et al. (2008) and Adkison et al. (2010) provide good examples of how to proactively investigate victim DDI potential of a BCRP substrate drug in a properly designed study (Table 2).
Summary of clinical studies using sulfasalazine as the intestinal BCRP probe
Recently, several null allelic mutations in the BCRP gene were reported to correlate with the Junior(a−) blood group antigen (Saison et al., 2012; Zelinski et al., 2012). The Junior(a−) phenotype has been associated with clinically relevant adverse events, including transfusion reactions and fetal anemia. This phenotype has been detected at a low frequency in individuals of northern European and Romani ancestry and in Bedouin Arabs, but at a relatively higher frequency in Asians, particularly Japanese (0.02%–1.7%) (Zelinski et al., 2012; Tanaka et al., 2014). All of these mutations result in nonfunctional protein, thereby identifying individuals with the Junior(a−) blood group as a population lacking BCRP activity.
As BCRP deficiency has recently been elucidated as the mechanism underlying the Junior(a−) phenotype, these populations have not yet been used to study BCRP-mediated drug disposition in humans. Initial investigations into the safety and efficacy implications of BCRP deficiency demonstrated conflicting results with respect to elevated blood concentrations of uric acid (no change and hyperuricemia) (Matsuo et al., 2009; Saison et al., 2012); however, uric acid disposition is complex and involves several transporters besides BCRP, and thus these conflicting findings are not entirely surprising (Reginato et al., 2012).
Theoretically, the Junior(a−) population may provide an opportunity to tease out the contribution of BCRP by comparing pharmacokinetics to healthy controls; however, such a clinical study paradigm remains to be established. In practice, these studies would have to be conducted in Japan, where the prevalence of the Junior(a−) phenotype is sufficiently high and the phenotype and study subject recruitment is enabled by tracking of this phenotype by blood banks.
Potential Clinical BCRP Probe Substrates
Based on studies with BCRP-transfected polarized cell lines and membrane vesicles generated from BCRP-overexpressing cells, many marketed drugs and dietary/natural constituents have been identified as substrates of BCRP. A list of 81 substrates was evaluated based on reported efflux ratios of >2 in cell monolayers and/or reported Km values of <10 μM (Table 3). These substrates have a broad range of physicochemical properties, with passive permeability ranging from low to high (0.07 × 10−6 to 21.8 × 10−6 cm/s). Many BCRP substrates are extensively metabolized, whereas others are excreted unchanged. The contribution of other transporters to the disposition of these substrates is common (Poirier et al., 2014). In contrast to the numerous BCRP substrates identified in vitro, the number of potential clinical substrates is limited. Theoretically, the ideal clinical probe substrate should be selective for BCRP, with minimal contribution from other transporters and metabolizing enzymes, to minimize the complexity in data interpretation and enable extrapolation to other BCRP substrate drugs. Analogous to digoxin safety studies for P-gp inhibitor drugs, if the candidate BCRP probe substrate has a narrow therapeutic window and/or identified safety concerns, then the DDI will have direct clinical relevance by informing dose adjustment for the substrate drug. An example of the latter is the case of statins, for which increased exposure can result in myopathy (Egan and Colman, 2011). Several potential clinical BCRP substrates for consideration are described below.
Compounds considered as in vivo human BCRP substrates based on available in vitro data
Metabolites are listed below the respective parent compound. Km and efflux ratio values are presented as reported in the respective references.
Sulfasalazine.
Sulfasalazine, an anti-inflammatory and immunomodulatory drug used to treat ulcerative colitis, rheumatoid arthritis, and Crohn’s disease (http://labeling.pfizer.com/ShowLabeling.aspx?id=524), has been proposed as a clinical probe for intestinal BCRP. Sulfasalazine is restricted largely to the gastrointestinal tract due to low permeability, low solubility, and efficient intestinal BCRP efflux. As such, oral bioavailability at therapeutic doses is low (<10%) and highly variable. The low fraction of a sulfasalazine oral dose that is absorbed into the systemic circulation is mostly excreted unchanged in urine (via glomerular filtration) and bile (http://labeling.pfizer.com/ShowLabeling.aspx?id=524; Giacomini et al., 2010).
Sulfasalazine is metabolized extensively by bacteria in the distal small intestine and colon to sulfapyridine, which is relatively well absorbed and cleared predominantly by N-acetyltransferase (NAT) 2. Inhibition of intestinal BCRP reduces sulfapyridine formation by increasing sulfasalazine absorption, which in turn decreases sulfasalazine exposure to intestinal bacteria. As such, the metabolite-to-parent AUC ratio is a more sensitive indicator of intestinal BCRP modulation in humans than parent sulfasalazine AUC, but only in intermediate and rapid acetylator phenotypes (Yamasaki et al., 2008). Specifically in NAT2 rapid acetylators, the range of BCRP clinical effect is ∼10-fold as measured by the decrease in sulfapyridine-to-sulfasalazine AUC ratio, while the clinical magnitude of parent sulfasalazine AUC increase is ≤3.5-fold (Yamasaki et al., 2008).
A wealth of preclinical data support sulfasalazine as an essentially specific probe for intestinal BCRP, although a potentially small contribution from P-gp and MRP2 has been proposed (Zaher et al., 2006; Dahan and Amidon, 2009; Zamek-Gliszczynski et al., 2012). Overall, Bcrp-knockout rodent data support BCRP limiting sulfasalazine oral bioavailability by at least an order of magnitude, while MRP2 and perhaps P-gp attenuate intestinal absorption by a lesser extent (≤2- to 3-fold). However, the fact that sulfasalazine intestinal absorption is not strictly limited by BCRP cannot be ignored in the interpretation of sulfasalazine interaction data, especially considering possible overlap between BCRP and P-gp, as well as BCRP and MRP2, inhibitors.
The possibility of uptake transporter(s) contributing to sulfasalazine absorption has been proposed, and intestinal OATP2B1 has been shown to transport sulfasalazine in vitro (Kusuhara et al., 2012; Tomaru et al., 2013). Intestinal uptake could be an important consideration concerning interpretation of DDI data should a decrease in sulfasalazine exposure be observed. Furthermore, saturation of uptake transporter(s) may underlie PK differences between subtherapeutic and therapeutic doses, which cannot be explained by drug solubility alone (Kusuhara et al., 2012).
Topotecan.
Topotecan is a topoisomerase inhibitor for second-line treatment of patients with metastatic ovarian carcinoma and small cell lung cancer, available in both intravenous and oral formulations (http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/200199s003lbl.pdf; http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020981s006lbl.pdf). Although oral absorption is limited by intestinal BCRP (Jonker et al., 2000; Kruijtzer et al., 2002), topotecan is a substrate for other efflux transporters as well, including P-gp and MRP2 (Lin et al., 2011). Nonetheless, in P-gp-knockout mice, the nonspecific BCRP and P-gp inhibitor elacridar increased topotecan oral exposure 6-fold, thus demonstrating a major role of Bcrp in limiting topotecan oral absorption (Jonker et al., 2000). Coadministration of elacridar with topotecan to cancer patients increased plasma exposure 2- to 3-fold (Kruijtzer et al., 2002), which is the upper limit of the possible increase in humans, where baseline bioavailability is 40% (http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020981s006lbl.pdf). The effect of the BCRP c.421C>A SNP on the PK of topotecan showed 1.3-fold-higher oral bioavailability in c.421C/A versus c.421C/C individuals, a modest effect that would be expected to increase in patients who are c.421A/A homozygous but were not included in this study (Sparreboom et al., 2005). Although topotecan has some attractive characteristics as a BCRP probe substrate, due to its safety profile as a cytotoxic agent, topotecan is impractical for routine use in healthy volunteer BCRP DDI studies.
Statins.
The physicochemical properties of statins, such as atorvastatin, cerivastatin, fluvastatin, pitavastatin, rosuvastatin, and pravastatin, are similar with respect to molecular weight, pKa, and anionic charge but differ in terms of lipophilicity. Consequently, the involvement of a variety of metabolizing enzymes and drug transporters in statin disposition differs based on a statin’s lipophilic versus hydrophilic character (Elsby et al., 2012; Shitara et al., 2013). Likewise, passive permeability varies with lipophilicity, and at one extreme is low for pravastatin and rosuvastatin while at the other is high for cerivastatin and fluvastatin (Jones et al., 2012; Menochet et al., 2012; Li et al., 2014). With respect to clearance pathways, cerivastatin, fluvastatin, simvastatin lactone, and atorvastatin are extensively metabolized, whereas pravastatin, rosuvastatin, and pitavastatin undergo minimal metabolism and are cleared mainly through biliary and urinary excretion (Elsby et al., 2012). All statins to a varying degree are substrates for hepatic uptake transporters, and some are BCRP substrates (Elsby et al., 2012).
Studies in Bcrp-knockout mice have demonstrated an important role for Bcrp in the hepatobiliary disposition of pitavastatin and rosuvastatin (Hirano et al., 2005; Kitamura et al., 2008). Following intravenous infusion to Bcrp-knockout mice, the biliary excretion rate of both drugs was reduced by approximately an order of magnitude relative to wild-type mice. In contrast, the systemic clearance of pitavastatin was not affected by Bcrp knockout, whereas that of rosuvastatin was reduced by ∼2-fold. Likewise, clinically, the BCRP c.421C>A polymorphism had a negligible effect on pitavastatin PK (Ieiri et al., 2007). Pitavastatin is a sensitive clinical probe for OATP1B1 DDI study (Prueksaritanont et al., 2014), but it is not recommended as a clinical probe in BCRP DDI study because of a minimal role of this transporter in its absorption, disposition, and clearance. Although the most pronounced effects on Cmax are reported for fluvastatin and rosuvastatin, the high permeability of fluvastatin combined with less compelling BCRP pharmacogenetic effects and DDI potential relative to rosuvastatin preclude consideration of this statin as a sensitive probe for intestinal BCRP-mediated DDI studies (Hirano et al., 2005; Ieiri et al., 2007; Elsby et al., 2012).
Rosuvastatin has limited ability to permeate cellular membranes via passive diffusion due to low lipophilicity (logD7.4 = −0.33) (Kitamura et al., 2008; Menochet et al., 2012). Intestinal absorption is ≤3.2-fold limited by BCRP in both preclinical species and humans (Karibe et al., 2014; Prueksaritanont et al., 2014). Rosuvastatin is primarily cleared by the liver (72%) and to a lesser extent by the kidneys (28%) (Elsby et al., 2012). Several hepatic uptake transporters (OATP1B1, OATP1B3, and sodium-taurocholate cotransporting polypeptide) contribute to its distribution in the liver, while BCRP and MRP2 secrete rosuvastatin into the bile, such that it can undergo extensive enterohepatic recirculation (Ho et al., 2006; Kitamura et al., 2008; van de Steeg et al., 2013). In addition to glomerular filtration, the tubular secretion of rosuvastatin is mediated by organic anion transporter 3 uptake and efflux by MRP2.
Based on pharmacogenetic assessment of all the statins, the disposition of rosuvastatin is affected primarily by BCRP polymorphisms in vivo, as summarized in Table 1. Because the BCRP c.421C>A variant does not represent a complete knockout of BCRP activity, it is likely that more profound changes may be observed if rosuvastatin was used in DDI studies with a potent BCRP inhibitor or in the Junior(a−) population, which lacks BCRP altogether. Moreover, reduced biliary transport of rosuvastatin via BCRP (due to either inhibition or a polymorphism) may lead to more pronounced changes in hepatic concentrations compared with systemic concentrations. Taken together, the evidence for BCRP as a major contributor to rosuvastatin disposition relative to the other statins is the strongest. However, the use of rosuvastatin as a BCRP DDI probe requires an appropriate clinical study design and careful consideration of perpetrator properties, as discussed below.
Clinical Evidence-Based Recommendations for BCRP Substrate Selection
Intestinal Probe Substrate.
Four clinical studies have employed oral sulfasalazine as an intestinal BCRP probe to investigate the PK effects of BCRP inhibitors and the c.421C>A SNP in humans (Urquhart et al., 2008; Yamasaki et al., 2008; Adkison et al., 2010; Kusuhara et al., 2012) (Tables 2 and 4). Although the number of human DDI studies is limited, important lessons can be drawn for future study design, specifically with respect to subject population, dose, formulation, and study endpoints. These considerations are discussed in detail below.
In vivo drug interaction studies involving rosuvastatin and sulfasalazine indicating at least partial contribution by BCRP
Population variability in sulfasalazine exposure can be large, as high as 81-fold (Adkison et al., 2010) (Table 4). As such, it is critical that clinical sulfasalazine interaction studies use a crossover design. To maximize the observed human intestinal BCRP interaction effects, study subjects with high BCRP and NAT2 activities should be enrolled, i.e., BCRP c.421C/C homozygous carriers and rapid NAT2 acetylators (Yamasaki et al., 2008). Rapid NAT2 acetylators provide the greatest sensitivity (∼10-fold) for determination of clinical DDIs in terms of the sulfapyridine-to-sulfasalazine AUC ratio, followed by intermediate acetylators (∼3- to 4-fold) versus slow acetylators, who show no apparent change in the ratio (Yamasaki et al., 2008). Notably, an unexpected increase in the sulfapyridine-to-sulfasalazine AUC ratio, or a decrease not commensurate with the increase in oral sulfasalazine exposure, may be indicative of NAT2 inhibition, which should be further investigated. In summary, intestinal BCRP DDI studies with sulfasalazine should enroll BCRP c.421C/C homozygous, rapid acetylator subjects and measure both parent and sulfapyridine oral exposure. As discussed in the preceding section, the sulfapyridine-to-sulfasalazine AUC ratio decrease appears to be a more sensitive measure of intestinal BCRP inhibition than parent drug exposure increase, and it is recommended that the DDI be reported using both endpoints.
The choice of sulfasalazine dose and formulation used for intestinal BCRP interaction studies also merits careful consideration (Table 2). BCRP pharmacogenetic and inhibition effects on oral sulfasalazine exposure were readily apparent and statistically significant following oral administration of a 2000-mg dose as immediate-release (IR) tablets (Yamasaki et al., 2008; Kusuhara et al., 2012). In contrast, following oral administration of 500-mg sulfasalazine enteric-coated tablets, systemic PK were not altered by BCRP pharmacogenetics or by oral coadministration of the BCRP inhibitor pantoprazole (Adkison et al., 2010). The exact mechanistic reasons for why sulfasalazine administered as enteric-coated tablets is not sensitive to BCRP modulation remain unknown. One plausible explanation is that this type of formulation releases drug farther down the gastrointestinal tract, where relative BCRP expression is up to 2-fold lower (Englund et al., 2006; Enokizono et al., 2007b; MacLean et al., 2008) and much less concentrated per unit of surface area (Pang, 2003), such that sulfasalazine is released past BCRP in the upper small intestine (Adkison et al., 2010). Although the extended-release formulation appears to be the most likely culprit for the lack of BCRP impairment effect in the Adkison et al. (2010) study, because three independent clinical studies using sulfasalazine IR tablets or suspension showed consistent PK changes (Urquhart et al., 2008; Yamasaki et al., 2008; Kusuhara et al., 2012), further work is needed to provide an unequivocal mechanistic explanation for the negative findings of Adkison et al. (2010). Other formulations have been evaluated, including an oral suspension prepared from crushed IR tablets. At a therapeutic dose of 1000 mg, oral exposure was 1.6-fold higher following administration of the suspension versus IR tablet but was not statistically significant (Urquhart et al., 2008; Kusuhara et al., 2012). However, following oral administration of a 0.1-mg suspension (microdose) versus a 2000-mg IR tablet, the dose-normalized exposure was an order of magnitude higher for the microdose suspension compared with the IR tablet, an effect possibly attributed primarily to potential saturation of the speculated intestinal absorptive transporter(s) at the therapeutic dose and secondarily to solubility effects (Kusuhara et al., 2012). Because the apparent fraction absorbed is greater for the microdose suspension, the magnitude of the intestinal BCRP DDI is not as large as that observed for the therapeutic IR tablet (AUC ratio of 2.0 versus 3.2) (Kusuhara et al., 2012).
Total daily doses of up to 2000 mg appear to be reasonably well tolerated in most subjects; however, gastric side effects are common, with about one-third of subjects experiencing these side effects to some degree and increasing with dose level (http://labeling.pfizer.com/ShowLabeling.aspx?id=524; Kusuhara et al., 2012). Serious side effects, such as Stevens-Johnson syndrome, can occur and have been observed following a single dose. To avoid unnecessary adverse event complications, DDI studies involving sulfasalazine should exclude subjects with known “sulfa” allergy, viral infections, a weakened immune system, a history or family history of Stevens-Johnson syndrome, or with the HLA-B*1502 allele. Based on currently available clinical experience, 1000 mg administered orally as IR tablets is the most optimal and practical sulfasalazine dosing regimen for intestinal BCRP DDI studies. Considering the maximal (<4-fold) clinical increase in sulfasalazine exposure due to BCRP inhibition, clinical exposure following administration of the 1000-mg dose would not exceed the exposure observed following administration of the highest approved dose level of 4000 mg/day. Finally, while the subject genotyping recommendations for robust response with sulfasalazine as a marker of intestinal BCRP activity (BCRP c.421C/C, NAT2 rapid acetylator phenotype, noncarriers of HLA-B*1502 allele) might seem onerous, these are all routine clinical genotyping tests, which add insignificant cost to the overall study and do not present a serious enrollment hurdle, because the predominant genotype is recommended for all three genes.
Systemic Probe Substrate.
Oral rosuvastatin represents a sensitive probe to assess the magnitude of inhibition of both intestinal and hepatic BCRP. Allowing for assessment of the worst-case DDI scenario, a rosuvastatin BCRP DDI study should enroll subjects homozygous for the BCRP reference allele c.421C/C (most prevalent in non-Asian ethnicities) and exclude any carriers of OATP1B1 c.521T/C or C/C. Consideration of CYP2C9 polymorphism (rosuvastatin undergoes minimal metabolism via this enzyme) is not of relevance because CYP2C9*1/*3 or *3/*3 was reported to have no effect on rosuvastatin (or its metabolite) PK or PD (Table 1). Alternatively, a clinical study should consider the genotype of the subjects enrolled and investigate the impact of multiple genetic covariates (e.g., coexistence of OATP1B1 and BCRP polymorphisms in the same subject) together with ethnicity data on the BCRP DDI sensitivity. An adequately powered study in terms of sample size would allow for identification of multiple high-risk combination patterns that could cause an increase in rosuvastatin exposure in conjunction with reduced BCRP activity, as recently reported for simvastatin (Tsamandouras et al., 2014).
Considering the complexity of rosuvastatin disposition and potential for enterohepatic recycling, it is evident that interpretation of increased rosuvastatin exposure in the DDI study following oral administration may be confounded by multiple factors. A key factor is the inability to differentiate between the contribution of intestinal and hepatic BCRP to the overall magnitude of a rosuvastatin-based interaction. Perpetrators that are potent BCRP as well as OATP inhibitors represent an additional issue of concern in that either or both can result in elevated blood levels of rosuvastatin. BCRP DDI study design involving oral and intravenous rosuvastatin formulations in the same subjects would allow delineation of the contribution of intestinal and hepatic BCRP. Based on the modeling and simulation work for compounds with comparable disposition to rosuvastatin (Watanabe et al., 2009; Chu et al., 2013b), the expectation is that the inhibition of hepatic BCRP may not cause significant changes in the plasma exposure, whereas drug accumulation in the hepatocytes and increase in liver exposure would occur. To account for this, the cholesterol-lowering effect of rosuvastatin will also be monitored in the control and inhibitor phase of the clinical studies, as a direct reflection of any changes in rosuvastatin liver exposure.
Rosuvastatin is commonly administered at an oral dose of 5–40 mg/day, with the majority of the clinical DDIs or pharmacogenetic studies conducted using a 10- or 20-mg dose (Simonson et al., 2004; Keskitalo et al., 2009b; Allred et al., 2011). Although a rosuvastatin intravenous formulation is not available currently, infusion of 8 mg rosuvastatin was reported as safe and well tolerated in a clinical study (Martin et al., 2003).
Rosuvastatin administered orally at a dose of 20 mg represents a good clinical probe for both hepatic and intestinal BCRP function (Table 1). The selected oral dose should be sufficient to capture BCRP DDI while minimizing any potential safety concerns associated with increased exposure at the upper limit of known clinical DDIs. Selection of a 20-mg oral rosuvastatin dose for the clinical DDI study can theoretically be paired with a 4-mg intravenous dose, reflecting the reported 20% oral bioavailability (Martin et al., 2003). In the case of intravenous rosuvastatin, inhibition of hepatic BCRP would interrupt enterohepatic recycling; monitoring of the changes in rosuvastatin PD effect relative to the control data would allow assessment of its contribution. Rosuvastatin DDI studies should be conducted in a crossover design, ideally in subjects with the highest BCRP and OATP activity, and report rosuvastatin PK and cholesterol-lowering effect at baseline and in the presence of a perpetrator. Comparison of the increase in rosuvastatin exposure after intravenous and oral administration and any changes in the PD effect will enable differentiation of the magnitude of hepatic and intestinal BCRP interaction.
Potential Clinical BCRP Inhibitors
Identifying a drug or diet-derived/natural product that can be used as a selective BCRP inhibitor in a clinical DDI study is challenging, as currently available compounds are not selective for BCRP and inhibit other transporters and drug-metabolizing enzymes (Leggas et al., 2006; Lainey et al., 2012; Fujita et al., 2013; Jamei et al., 2014; Poirier et al., 2014; Shukla et al., 2014). Upon careful consideration of inhibition data generated from various in vitro and in vivo studies, there is a high likelihood that any known BCRP inhibitor will inhibit other membrane transporters or metabolic enzymes. Given these limitations, the most potent and selective BCRP inhibitors that complement the proposed BCRP substrates were identified for their potential use in clinical DDI studies. Such inhibitors were selected through a comprehensive literature search using the University of Washington Drug Interaction Database (http://www.druginteractioninfo.org) and the University of California, San Francisco-FDA TransPortal (http://bts.ucsf.edu/fdatransportal).
Identifying Potential BCRP Inhibitors.
Known BCRP inhibitors with IC50 (or Ki) values of <10 μM were identified that could potentially be used in a clinical DDI study (Table 5). Compounds for which the IC50 could not be obtained from the literature or considered impractical for use in a clinical study (e.g., cyclosporine) were not considered further. Cmax values for the identified BCRP inhibitors were used to determine [I1]/IC50 values, which were then used to predict the potency of systemic BCRP inhibition (US Food and Drug Administration, 2012; European Medicines Agency, 2013). Similarly, maximum intestinal concentrations, estimated as the highest approved dose/250 ml, were used to calculate [I2]/IC50 values, which were used to predict the potency of intestinal BCRP inhibition (Supplemental Table 1) (US Food and Drug Administration, 2012; European Medicines Agency, 2013). If only a Ki was available, the IC50 was assumed to be equivalent to 2 × Ki when substrate concentration is equal to the Km (Zhang et al., 2008). Drugs for which literature data supported inhibition of BCRP in vivo were included in the final assessment of potential inhibitors of systemic and intestinal BCRP. To undergo further consideration, a compound had to meet our criteria set at [I1]/IC50 > 1 or [I2]/IC50 > 20 for a high likelihood of clinical inhibition, which had to be supported by literature evidence that the compound either inhibits BCRP or is a BCRP substrate in vivo (i.e., a competitive inhibitor). BCRP inhibitors were rank ordered from highest to lowest [I1]/IC50 (Table 6) and [I2]/IC50 (Table 7).
Compounds considered as in vivo BCRP inhibitors, based on in vitro IC50 or Ki values <10 μM
IC50 and Ki values are presented as reported in the respective references.
In vitro and in vivo data and considerations for potential systemic BCRP inhibitors
IC50 and Ki values are presented as reported in the respective references, along with the substrate used.
In vitro and in vivo data and considerations for potential intestinal BCRP inhibitors
IC50 and Ki values are presented as reported in the respective references, along with the substrate used.
In addition to a rank ordering of systemic and intestinal BCRP inhibition potential, a detailed assessment of the inhibition potential of other transporters and drug-metabolizing enzymes was considered (Tables 6 and 7). The comprehensiveness of these tables reflects the difficulty encountered when selecting a viable substrate-inhibitor pair, as multiple inhibitory interactions occurring simultaneously will confound data interpretation concerning specific BCRP involvement in substrate PK. Such potential nonselective interactions were carefully considered, after which most identified BCRP inhibitors were eliminated. The final selected BCRP inhibitors have inherent pros and cons, which are detailed below.
Sulfasalazine.
Sulfasalazine is a BCRP substrate with limited absorption due to efficient intestinal BCRP efflux into the lumen, as well as low permeability and low solubility (http://labeling.pfizer.com/ShowLabeling.aspx?id=524). Being a substrate, sulfasalazine may act as a competitive inhibitor of BCRP, with reported IC50 values as low as 0.46 μM (Elsby et al., 2011). The Cmax range for a single 3- to 4-g dose of sulfasalazine is 38–79 μM (Goodman et al., 2001), resulting in a maximum [I1]/IC50 value of 170 (Table 6) and an [I2]/IC50 value of 87,000 (Table 7). As such, sulfasalazine could be used as a BCRP inhibitor in a clinical study when paired with a BCRP substrate such as rosuvastatin. However, sulfasalazine also inhibits OATP-mediated uptake, in particular OATP1B1 (IC50 = 0.56 μM) (Karlgren et al., 2012b). As OATP1B1 is one of the primary transporters responsible for taking up rosuvastatin into the liver (Simonson et al., 2004; Ho et al., 2006; Pasanen et al., 2007), and the inhibitory potency toward OATP1B1 is similar to that of BCRP, OATP1B1 becomes a major confounding factor. Therefore, sulfasalazine was excluded from consideration for use as a clinical BCRP inhibitor.
Elacridar.
Elacridar was originally designed and developed as a reversal agent of P-gp and BCRP to overcome multidrug resistance. Although elacridar increases the accumulation of several anticancer agents in preclinical tumor models (Hyafil et al., 1993; Agarwal et al., 2012), it is not a systemically potent efflux inhibitor at chronically tolerated doses in humans (Kalvass et al., 2013). However, in the intestine, elacridar has been shown to increase topotecan bioavailability via inhibition of BCRP (Kruijtzer et al., 2002; Kuppens et al., 2007). These observations are consistent with elacridar ranking low in terms of systemic BCRP inhibition ([I1]/IC50 = 1.1) (Table 6), but high with respect to intestinal BCRP inhibition ([I2]/IC50 = 9200) (Table 7). The use of elacridar as a clinical intestinal BCRP inhibitor is precluded by the fact that this compound is not currently approved by the FDA or EMA for use in humans. Furthermore, elacridar is an even more potent inhibitor of P-gp than BCRP ([I2]/IC50 = 180,000) and would not be suitable for elucidating the intestinal BCRP contribution to limiting the absorption of dual substrates.
Proton Pump Inhibitors.
Multiple proton pump inhibitors (PPIs) inhibit BCRP in vitro; however, pantoprazole and rabeprazole are the only two that meet the aforementioned criteria for consideration as clinical inhibitors (Tables 6 and 7); however, clinical and/or preclinical proof-of-concept studies have not been reported. Despite clear advantages to using an inhibitor with an acceptable safety profile such as pantoprazole (Remes-Troche et al., 2014) or rabeprazole (Zouboulis-Vafiadis et al., 2014), these drugs are not as potent BCRP inhibitors as the tyrosine kinase inhibitors (TKIs) with respect to the [I2]/IC50 values (Table 7), for which preclinical proof of concept has been demonstrated (Zaher et al., 2006). Similarly, pantoprazole marginally meets the predefined criterion for systemic BCRP inhibition, with an [I1]/IC50 value of 1.2 (Table 6). As such, although using a PPI may minimize the safety risks, other drugs are expected to offer a superior in vivo inhibition profile, rendering data interpretation more straightforward. Finally, any observed DDIs with PPIs are fundamentally confounded by increased gastrointestinal pH (Ware et al., 2013), and a separate intestinal pH control arm must be incorporated into the clinical study with a PPI that is not a BCRP inhibitor to control for this effect (Adkison et al., 2010).
Natural Products.
Several diet-derived/natural product constituents, such as curcumin, daidzein, genistein, chrysin, 7,8-benzoflavone, and glycyrrhetic acid, have been shown to inhibit human BCRP in vitro (Zhang et al., 2005a; Yoshida et al., 2008; Merino et al., 2010; Tamaki et al., 2010). In addition, a mixture of the soy isoflavones genistein and daidzein was shown to inhibit Bcrp in wild-type but not Bcrp-knockout mice, supporting specificity for Bcrp inhibition (Merino et al., 2010). Of these constituents, only curcumin has been evaluated clinically as a BCRP inhibitor. Curcumin is the principal curcuminoid in turmeric, a commonly used spice derived from the rhizomes of Curcuma longa, a member of the ginger family. Curcuminoids are polyphenols that render turmeric yellow in color (Sharma et al., 2005). Curcumin is a potent inhibitor of BCRP, with a Ki of 0.7 μM determined with membrane vesicles overexpressing human BCRP and sulfasalazine as the probe substrate (Kusuhara et al., 2012). Based on the [I2]/IC50, curcumin is predicted to inhibit intestinal BCRP in vivo. Several clinical studies involving oral curcumin have been conducted, with doses ranging from 2000–8000 mg (Cheng et al., 2001; Kusuhara et al., 2012). Curcumin was well tolerated, with no signs of toxicity observed during a phase 1 study in which the subjects were administered oral doses of 8000 mg/day for 3 months (Cheng et al., 2001). At 2000 mg/day, curcumin increased sulfasalazine exposure 3.2-fold, which was attributed to inhibition of intestinal BCRP (Kusuhara et al., 2012). Because curcumin is considered a food or dietary supplement, and thus not subject to strict manufacturing controls as drugs, the purity and reproducibility with respect to constituent composition, stability, and dissolution characteristics is not assured. Accordingly, a natural products chemistry laboratory should be consulted for sourcing and analyzing the test product.
TKIs.
TKIs are a relatively new generation of anticancer drugs that are generally well tolerated. Several TKIs (erlotinib, gefitinib, lapatinib, nilotinib, and sunitinib) are potent BCRP inhibitors with acceptable safety margins to be considered for use in healthy volunteers. One clear advantage to using a TKI as a clinical BCRP inhibitor is that, with the exception of sunitinib, TKIs are predicted to inhibit intestinal as well as systemic BCRP. Based on the rank ordering of inhibitory potency for TKIs, lapatinib is the most potent when considering both intestinal and systemic inhibition of BCRP. Although in vitro data suggest that lapatinib may inhibit P-gp, OATP1B1, and CYP2C8 concomitantly, these interactions are based on a high therapeutic dose of 1250 mg. However, at a subtherapeutic dose of 250 mg, based on [I1]/IC50 and [I2]/IC50 values (22 and 40,000, respectively), lapatinib is predicted to effectively inhibit BCRP (Tables 6 and 7). Although lapatinib carries a black box warning for hepatotoxicity rarely observed during chronic use in cancer patients (http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022059s016s017lbl.pdf), it is well tolerated acutely, as doses up to 7000 mg/day have shown no dose-limiting toxicities (Chien et al., 2014). At clinical doses, the diarrhea associated with patient treatment would be the only potential concern; however, this adverse effect typically resolves within 24–48 hours of treatment cessation (MacFarlane and Gelmon, 2011; Chien et al., 2014).
Clinical Evidence–Based Recommendations for BCRP Inhibitor Selection
Considering the totality of available information, two compounds emerge as potential clinical BCRP inhibitors for use in DDI studies. To discern the effect of intestinal-specific BCRP inhibition, a single 2000-mg dose of curcumin is proposed to extensively inhibit intestinal, but not systemic, BCRP (Tables 6 and 7). Due to poor absorption, curcumin is not detectable in plasma at this proposed dose (Garcea et al., 2005; Kusuhara et al., 2012), rendering the compound a practical choice for intestinal-specific BCRP inhibition. The second proposed BCRP inhibitor is lapatinib. Compared with a food product such as curcumin, the benefit of using a marketed drug to assess DDIs is that, among other things, a natural products chemistry laboratory need not be consulted. The recommended subtherapeutic oral dose of 250 mg should have minimal adverse effects and possibly attenuate the impact of inhibition of other transporters and metabolic enzymes (Castellino et al., 2012). At this dose, lapatinib is predicted to inhibit both intestinal and systemic BCRP based on [I1]/IC50 and [I2]/IC50 values (Tables 6 and 7).
One potential concern is that lapatinib may also inhibit OATP1B1, as rosuvastatin uptake is dependent on this transporter (the [I1]/IC50 is 0.29, which is marginally above the cutoff value of 0.1). However, lapatinib is highly protein bound (>99%), so circulating levels of free drug available to inhibit OATP1B1 systemically will be significantly less than the total Cmax reported in Table 6. Taking into account the concentration of lapatinib in the intestine and in the systemic circulation, lapatinib is likely to inhibit BCRP but not OATP1B1.
Recommendations for Clinical BCRP Drug Interaction Study Design
This comprehensive review of potential clinical BCRP probe substrates and inhibitors identified two substrates and two inhibitors to assess BCRP-mediated DDIs in humans. The two recommended probe substrates are sulfasalazine (1000 mg, IR, oral) and rosuvastatin (20 mg, oral; and 4 mg, i.v.), and the two inhibitors are curcumin (2000 mg, oral) and lapatinib (250 mg, oral). The combination of oral sulfasalazine with either inhibitor provides a substrate-inhibitor pair to assess the extent of a potential intestinal BCRP DDI. The dynamic range of sulfasalazine will provide a measure of the highest extent of an interaction due to inhibitor coadministration. Further, the combination of oral and intravenous rosuvastatin with both inhibitors will enable deconvolution of intestinal versus systemic BCRP contribution to rosuvastatin PK. Based on these aims, a clinical study design is proposed to validate these approaches (Fig. 1).
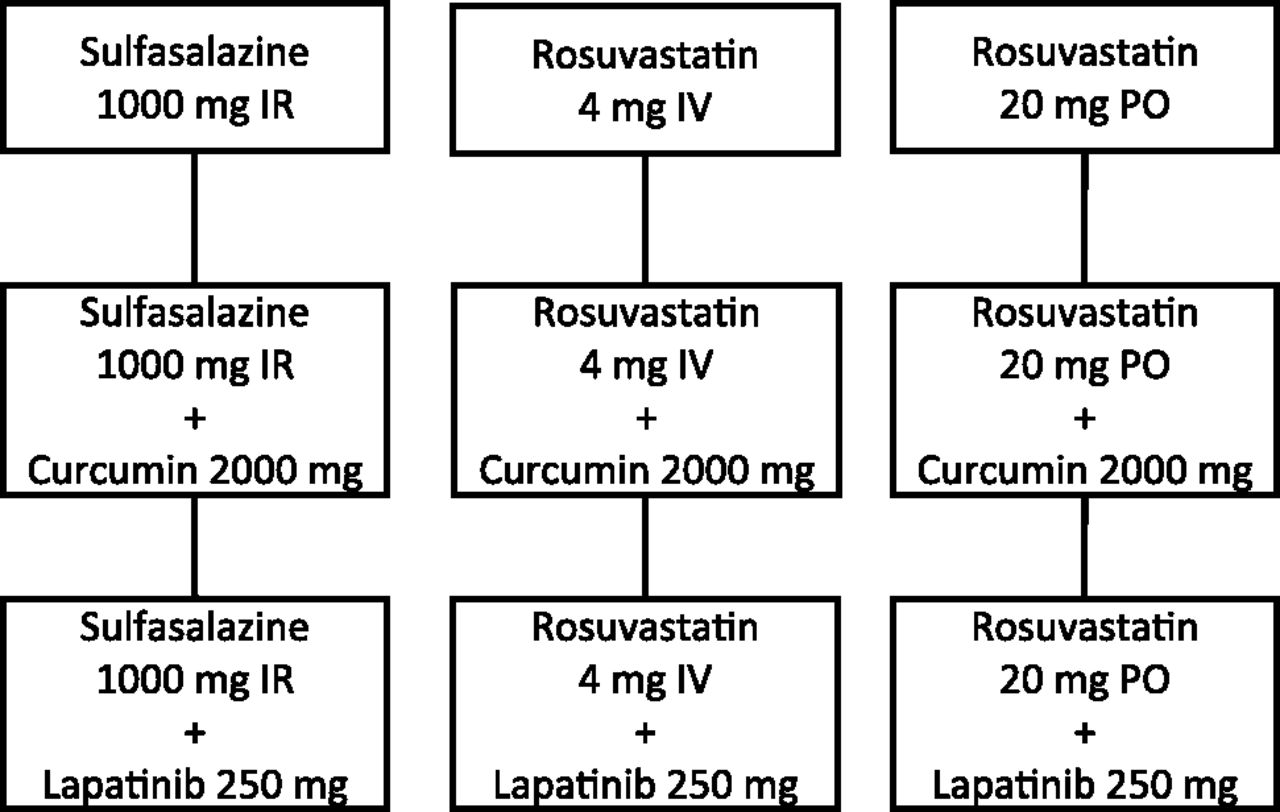
Outline of proof-of-concept clinical study to validate the proposed BCRP DDI evaluation approach. Clinical dosing scheme for oral (PO) immediate release (IR) sulfasalazine alone and with PO curcumin or PO lapatinib, intravenous (IV) rosuvastatin alone and with PO curcumin or PO lapatinib, and PO rosuvastatin alone and with PO curcumin or PO lapatinib.
Sulfasalazine Intestinal BCRP DDI.
Oral sulfasalazine (1000 mg, IR, single dose) is recommended to evaluate intestinal BCRP-mediated DDIs. The study will be an open-label, three-period crossover study involving 10 subjects genotyped for the BCRP reference allele and phenotyped for rapid NAT2 acetylator status prior to enrollment. Sulfasalazine will be administered alone and in combination with oral curcumin (2000 mg, single dose) or lapatinib (250 mg, single dose) (Fig. 1). Subjects will undergo three treatments with at least a 1-week washout between treatments as follows: sulfasalazine alone, sulfasalazine plus curcumin, and sulfasalazine plus lapatinib. PK of both sulfasalazine and sulfapyridine will be determined, with the DDI reported as both an increase in sulfasalazine exposure and decrease in the sulfapyridine-to-sulfasalazine AUC ratio.
Rosuvastatin Intestinal and Hepatic BCRP DDI.
Oral rosuvastatin (20 mg, single dose) is recommended to evaluate intestinal and hepatic BCRP DDIs, whereas intravenous rosuvastatin (4 mg, single dose) is recommended to evaluate hepatic BCRP DDIs. Each rosuvastatin administration route will include an open-label, three-period crossover study involving subjects genotyped for the BCRP and OATP1B1 reference alleles prior to enrollment. Rosuvastatin will be administered alone and with oral curcumin (2000 mg, single dose) or lapatinib (250 mg, single dose) (Fig. 1). Treatments include intravenous rosuvastatin alone, intravenous rosuvastatin plus oral curcumin, intravenous rosuvastatin plus oral lapatinib, oral rosuvastatin alone, oral rosuvastatin plus oral curcumin, and oral rosuvastatin plus oral lapatinib. Inclusion of cholesterol monitoring will provide insight into hepatic accumulation of rosuvastatin due to inhibition of hepatic BCRP. Changes in rosuvastatin exposure relative to the control will be the DDI study endpoint.
Conclusions
The regulatory recommendation to evaluate the contribution of BCRP to drug disposition and DDIs in clinical studies has posed challenges across the pharmaceutical industry. These challenges primarily reflect the lack of selective and specific human BCRP substrates and inhibitors, as well as a lack of established and accepted optimal clinical study designs. As described throughout this review, BCRP substrates and inhibitors suitable for human use were identified through a comprehensive analysis of available pharmacogenetics and DDI data. Two substrates were selected to probe intestinal and/or hepatic BCRP-mediated DDIs. Oral sulfasalazine (1000 mg, IR tablets) was deemed the best available probe for intestinal BCRP, oral rosuvastatin (20 mg) as the best available probe for both intestinal and hepatic BCRP, and intravenous rosuvastatin (4 mg) as the best available probe for hepatic BCRP. Oral curcumin (2000 mg) and oral lapatinib (subefficacious 250-mg dose) were deemed the best available BCRP inhibitors. Optimal study designs that would elicit the worst-case clinical BCRP DDI scenarios are proposed (Fig. 1). The PK data obtained from the proposed nine-arm clinical study should provide the pharmaceutical industry and regulatory agencies insight into the extent of BCRP-mediated clinical DDIs, as well as validate clinical study designs that enable optimal assessment of intestinal and combined intestinal and hepatic BCRP-mediated DDIs.
Authorship Contributions
Participated in research design: Lee, O’Connor, Galetin, Cook, Ellens, Feng, Taub, Paine, Polli, Ware, Zamek-Gliszczynski.
Performed data analysis: O’Connor, Ragueneau-Majlessi, Ritchie.
Wrote or contributed to the writing of the manuscript: Lee, O’Connor, Ritchie, Galetin, Cook, Feng, Ellens, Ragueneau-Majlessi, Taub, Paine, Polli, Ware, Zamek-Gliszczynski.
Footnotes
- Received November 16, 2014.
- Accepted January 9, 2015.
↵1 Current affiliation: Drug Metabolism and Pharmacokinetics, Ardea Bioscience Inc., San Diego, California.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the concentration-time curve
- BCRP
- breast cancer resistance protein
- DDI
- drug-drug interaction
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- IR
- immediate release
- MRP
- multidrug resistance–associated protein
- NAT
- N-acetyltransferase
- OATP
- organic anion–transporting polypeptide
- PD
- pharmacodynamics
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- PPI
- proton pump inhibitor
- SNP
- single nucleotide polymorphism
- TKI
- tyrosine kinase inhibitor
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Clinical Relevance of BCRP Polymorphisms
- Potential Clinical BCRP Probe Substrates
- Clinical Evidence-Based Recommendations for BCRP Substrate Selection
- Potential Clinical BCRP Inhibitors
- Clinical Evidence–Based Recommendations for BCRP Inhibitor Selection
- Recommendations for Clinical BCRP Drug Interaction Study Design
- Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI