


Abstract
To examine the possibility for drug metabolism polymorphism, adult human flavin-containing monooxygenases (form 3) (EC 1.14.13.8) that differ at one amino acid were expressed in Escherichia colias maltose binding protein fusions. The cDNA that was first reported during the cloning of adult human flavin-containing monooxygenase was designated the wild type lys158 enzyme. A second cDNA has been identified as a common polymorphism in some human populations and was designated the glu158enzyme. The cDNA that encodes both enzymes was subcloned into a high yield protein fusion expression system, expressed, and the protein was partially purified by affinity chromatography and characterized for enzyme activity with selective functional substrate probes.N- and S-oxygenation activity of both enzymes was determined with 10-(N,N-dimethylaminopentyl)-2-(trifluoromethyl)phenothiazine and methyl p-tolyl sulfide, respectively. It was found that expression of both lys158 and glu158 enzymes of the human flavin-containing monooxygenase form 3 as fusions with the maltose binding protein resulted in an enzyme that was soluble and greatly stabilized and had a reduced requirement for detergent during enzyme purification and during the assay for activity. Expression of the fusion proteins has allowed the preparation of stable and highly active enzyme at greater purity than was readily possible in the past. With the exception of the stability and solubility characteristics, the physical and chemical properties of lys158 and glu158 maltose binding fusion proteins of human flavin-containing monooxygenase form 3 variants resembled that of flavin-containing monooxygenase enzyme activity associated with human liver microsomes and enzyme isolated from a previous Escherichia coli expression system that lacked the protein fusion. Comparison of the catalytic activity of the two fusion proteins showed that while both forms were active, there were differences in their substrate specificities. Expression of the adult human flavin-containing monooxygenase form 3 as a maltose binding protein has allowed considerable advances over the previously reported cDNA-expressed enzyme systems and may provide the basis for examining the role of the flavin-containing monooxygenase in human xenobiotic or drug metabolism.
The flavin-containing monooxygenase (FMO) consists of a family of enzymes involved in the metabolism of drugs and endogenous chemicals (1, 2). In humans, the flavin-containing monooxygenase form 3 (FMO3)1 (EC 1.14.13.8), is the predominant form of flavin-containing monooxygenase expressed in adult human liver (3, 4). cDNA-expressed human FMO3 (5-7) and human liver microsomal FMO3 (8-15) have been observed to N- andS-oxygenate nucleophilic nitrogen- and sulfur-containing drugs and chemicals, respectively. The physiological role of human FMO3 in hepatic metabolism is not clear, but it has been suggested that human FMO3 participates in the detoxication of nucleophilic heteroatom-containing dietary, endogenous or xenobiotic chemicals to produce polar, oxygenated metabolites that are readily excreted (16).
Human FMO3 is apparently responsible for the conversion of trimethylamine (TMA) to trimethylamine N-oxide (TMANO) in normal individuals (17, 18), although this has never been directly demonstrated with substantially pure human FMO3 (1). Human subjects that do not convert at least 65% of an administered dose of TMA to TMANO suffer from trimethylaminuria (17, 19-21). Trimethylaminuria patients excrete relatively large amounts of TMA in their urine, sweat, and breath and exhibit a fishy odor characteristic of TMA, and this is why the patients have been designated as having the “fish-odor” syndrome (17-22). Evidence has been presented that the parents of affected individuals had impaired ability to N-oxygenate amine substrates including nicotine, nicotinamide, guanethidine, and metyrapone (17, 23). Although Prader-Willi syndrome has been postulated to be associated with trimethylaminuria (24), Prader-Willi syndrome is known to be associated with a deletion of a normal allele located in the 15q11-q13 region of the chromosome (24), and this location is distinct from the position observed for human FMO genes (i.e. chromosome 1) (25) that has been postulated to lead to the expression of a recessive gene for defective human FMO. While a defective human FMO has been postulated to be responsible for trimethylaminuria, to date the functional consequence for a mutant human FMO has not been reported.
Recently, selective functional probe substrates have been used to examine a role of human FMO3 in adult human liver metabolism. In vitro/in vivo correlations have shown that stereoselective (S)-nicotine N-1′-oxygenation and cimetidine S-oxygenation are useful indicators of human FMO3 activity (6-8), and in vitro regioselectiveN-oxygenation of 10-(N,N-dimethylaminoalkyl)phenothiazines are useful indicators of animal FMO activity (26). Undoubtedly, many additional examples of selective functional substrates for human FMO3 will be discovered. Consequently, knowledge of expression of human FMO3 enzymes showing polymorphism for drug or endogenous chemical metabolism will be an important consideration.
Herein, we report the kinetic comparison of the wild type and a variant of human FMO3 fusion proteins that were discovered in the human population from sequencing and restriction length polymorphism studies.2 The cDNA-expression, purification and enzymatic assay and comparison of the physical chemical and catalytic properties for the two human FMO3 enzymes examined was facilitated by the adoption of a protein fusion expression system. The expressed human FMO3 fusion proteins were isolated in a soluble form that significantly reduced the requirement for added detergent. The two human FMO3 enzymes examined showed distinct substrate specificities. Variation at the locus that encodes human FMO3 may represent a predisposition of certain populations to altered drug or chemical metabolism and hence altered clearance parameters. Human polymorphism for oxygenation of nucleophilic heteroatom-containing chemical may contribute to unusual human drug metabolism reactions that may lead to drug-drug interactions that could in turn contribute to idiosyncratic disease conditions (27).
Materials and Methods
Chemicals.
Triton X-100, L-α-phosphatidylcholine, and the components of the NADPH-generating system were purchased from Sigma Chemical Co. (St. Louis, MO). Methyl p-tolyl sulfide, (trifluoromethyl)phenothiazine, dicyclohexylcarbodiimide (DCC), and all other chemicals used in this study were purchased from Aldrich Chemical Co. (Milwaukee, WI). All other reagents and solvents used in this study were purchased from Fisher Scientific (Santa Clara, CA). The pGEM vector, restriction endonucleases, DNA polymerase Klenow fragment, T4 DNA ligase, and phosphatase and Taq polymerase were obtained from Promega (Madison, WI). Mutagenesis reagents were from the Muta-Genein vitro mutagenesis kit from Bio-Rad (Hercules, CA). Oligonucleotides for mutagenesis and sequencing were synthesized by DNagency (Malvern, PA). Vector pMAL-c2 and amylose resin were purchased from New England Biolabs (Beverly, MA). Sephacryl 300 was obtained from Pharmacia (Piscataway, NJ.).
Synthesis.
10-(N,N-Dimethylaminopentyl)-2-(trifluoromethyl)phenothiazine (5-DPT) was synthesized by a modification of the procedure previously described (26). 10-(N,N-Dimethylaminopentyl)-2-(trifluoromethyl)phenothiazineN-oxide (5-DPTNO) was synthesized and characterized spectroscopically as previously described (5). Methylp-tolyl sulfoxide (MTSO) was synthesized from methylp-tolyl sulfide (MTS) according to the general method of Cashman et al. (8, 9).
Oligonucleotide Sequencing.
Oligonucleotide sequencing was done by the DNA sequencing facility at the Seattle Biomedical Research Institute (Seattle, WA) using Sanger sequencing technology. Automated electrophoresis and data recording were done with an Applied Biosystems Sequencer Model 373A (Foster City, CA).
Subcloning Human FMO3 cDNA into the Maltose Binding Protein Fusion Expression System.
Two oligonucleotides were synthesized to use in PCR amplification of the human FMO3 cDNA (3). Proper insertion of human FMO3 cDNA into the expression vector pMAL-c2 and PCR amplification was done in a way that allowed the fusion of human FMO3 cDNA at the 3′ end of sequences encoding the maltose binding protein (MBP). Oligonucleotides that corresponded to 5′-GGGAAGAAAGTGGCCATC-3′ and 5′-CCGGTCGACGGATCCAAGCTTAGGTCAACACAAGG-3′ were used as the 5′ and 3′ PCR primers, respectively. The 3′ oligonucleotide includedHindIII, BamHI, and SalI sites that allowed, among other manipulations, insertion of the PCR fragment into the pMAL-c2 vector between the Xmn I site (blunt end) and the HindIII site.
The two oligonucleotides were used in PCR amplification reactions with the full length human FMO3 cDNA-containing vector (i.e.pHFMO3-f1). Approximately 0.1 μg of the single-stranded form of pHFMO3-f1 was used as the template for the PCR. One hundred pmol of each primer was used in a 50-μl reaction under standard PCR buffer conditions. PCR cycle conditions were 94°C melting (1 min), 55°C annealing (1 min), and 74°C extension (1.5 min), and after 20 cycles yielded sufficient DNA to proceed with the cloning steps. The PCR product was treated with DNA polymerase I (Klenow fragment) to ensure that the DNA fragments had flush ends prior to cloning. The successful result of cloning the human FMO3 cDNA PCR fragment into theXmn I and HindIII sites of the vector pMAL-c2 gave a construct that was named pMAL-HFMO3. Several clones were examined for expression in JM109 E. coli before one was isolated that produced a product with the expected size of the full length fusion protein (i.e., 100 kDa).The junction between the maltose binding protein (MBP) and human FMO3 contained a Factor Xa cleavage site that was a feature of the pMAL-c2 vector. The fusion junction was designed so that cleavage of the human FMO3-MBP fusion protein with Factor Xa could release a human FMO3 product that contained an amino-terminal glycine that corresponded to the FMO3 enzyme from human liver (3).
Mutagenesis of Human FMO3 cDNA Encoding Lysine 158: Conversion to Glutamate 158.
Herein we report that there are two human FMO3 enzymes that have been identified and expressed.2The lys158 codon has been designated as the wild type and glu158 has been designated as a variant. The lysine 158 codon of human FMO3 cDNA is AAG. A single oligonucleotide base change of G converts the lys 158 codon to one for glutamate (i.e. GAG). The oligonucleotide base change was accomplished by site-directed mutagenesis using the BioRad Mutagene kit. The oligonucleotide synthesized for this purpose was: 5′-CC TGG AAA GGA CT(G/C) TTT TGG TAG GTT GGG-3′. The above oligonucleotide was designed to change lysine 158 to either a glutamate or glutamine. In several attempts to express the variants, only the clone encoding the glutamate amino acid was observed. This result was verified by oligonucleotide sequencing of the entire coding region of the clones as well as part of the cDNA encoding the MBP. The mutagenesis was carried out on a subclone of human FMO3 cDNA (i.e., NcoI to SacI fragment) in vector pGEM(-). The cDNA encoding the glutamate variant was then transferred to the MBP-fusion expression vector by exchanging the NheI to SacI fragment of the mutagenized vector with that of vector pMAL-HFMO3.
Expression of the human FMO3 MBP fusion protein and purification with Amylose Resin.
Starter cultures of JM109 E. coli containing pMAL-HFMO3 were grown overnight on selective SOC medium (50 μg/ml ampicillin) (28). A fresh overnight culture was diluted 100-fold in SOC medium in the presence of 100 μg/ml ampicillin. The culture was grown at 37°C for 2 hr with vigorous shaking. After 2 hr, 8 mg/l riboflavin was added and the cultures were induced with the addition of isopropyl β-D-thiogalactopyranoside (IPTG) to achieve a final concentration of 0.6 mM. The cultures were incubated for an additional 3 hr at 37°C with vigorous shaking. At 3 hr the cells were removed from the incubator and chilled on ice. In general, fusion protein preparations provided protein with equal efficiency on several scales (i.e. 25 ml, 250 ml, and 4 liter). A 4-liter culture was harvested at 2–4°C by centrifugation at 6,000g for 15 min, washed once with 400-ml 50 mM sodium phosphate buffer, pH 8.4, and resuspended in 80 ml of lysis buffer (i.e. 50 mM sodium phosphate, pH 8.4, 0.1% lecithin, 0.5% triton X-100, 0.5 mM phenylmethylsulfonylfluoride (PMSF)) (29). The cell suspension was then allowed to freeze slowly at −20°C. After freezing, the cells were either transferred to −80°C for storage or thawed prior to sonication. The freeze-thaw process appeared to improve the efficiency of lysis. Sonication was done in three bursts (60 sec, 30 sec, and 20 sec for the 4-liter preparations) with a Braun Sonic Model 2000 Sonicator fitted with a microprobe. The bacterial lysates were cleared by centrifugation at 14,000g for 20 min to provide a supernatant.
The supernatant derived from a 4-liter culture of JM109 E. coli containing pMAL-HFMO3 was passed through an amylose column once that was pre-equilibrated in column buffer (50 mM sodium phosphate buffer, pH 8.4). The amylose affinity column was then rinsed with 10 column volumes of column buffer that was sufficient to remove essentially all unbound proteins. Proteins that were bound by virtue of their affinity to amylose were then released with column buffer that contained 10 mM maltose. Fractions were collected and assayed for protein concentration and fractionated on SDS-PAGE. As described previously, for non-fusion protein human FMO3, selective precipitation with PEG 8000 afforded more highly purified fractions (5, 29). Generally, 86% and 81% of the activity of human glu158 and lys158 FMO3 MBP, respectively, was recovered using this procedure.
Fractionation of human FMO3 MBP on Sephacryl S-300.
As judged by 5-DPT N-oxygenation, the most active fraction from a representative amylose affinity chromatography fractionation were pooled (approximately 2.6 mg of protein) and placed on a size exclusion chromatography Sephacryl S-300 column equilibrated in 50 mM sodium phosphate buffer, pH 8.4, and 0.5% Triton X-100. One ml fractions were collected and further analyzed for 5-DPTN-oxygenation activity and protein concentration and by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS PAGE) (data not shown). Protein concentration was determined with the Pierce BCA method (Rockford, IL).
Factor Xa Cleavage.
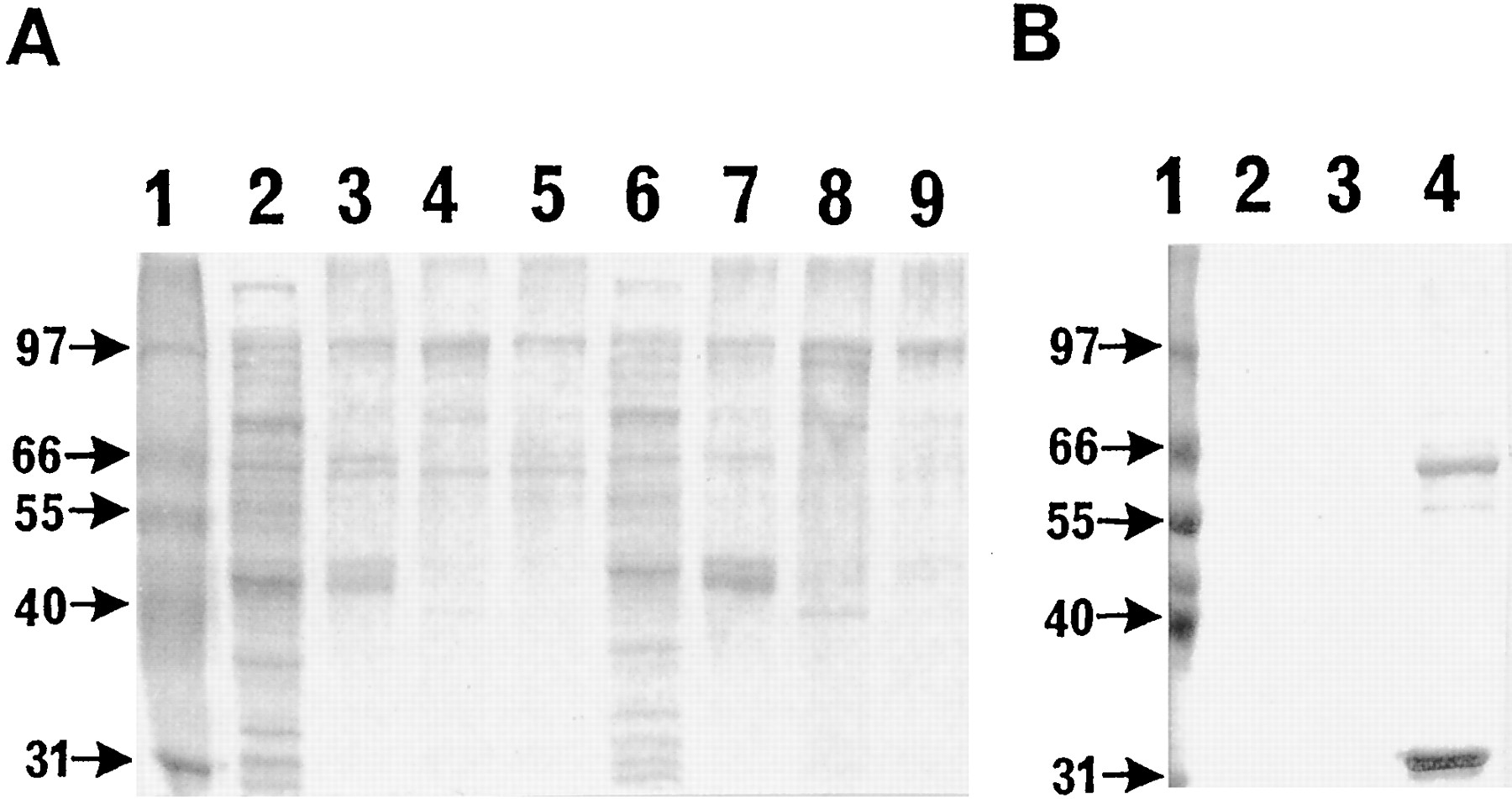
Two preparations of human lys158 FMO3 MBP were used in the Factor Xa cleavage reaction. The 14% PEG 8000 precipitate (fig. 1A) and a fraction of lys158 FMO3 MBP from a purification by Sephacryl S-300 chromatography (both 0.4 mg/ml) were also used in a Factor Xa cleavage reaction. Typically, 24 μg of lys158FMO3 MBP was placed in a total of 100 μl of 100 mM sodium phosphate buffer (pH 7.6), and 7.6 μg of Factor Xa was added (a gift from Dr. K Kujikawa and Professor E. Davie, University of Washington) and incubated at 37°C for 1 or 3 hr. Fig. 1B shows the reaction product of the Factor Xa cleavage of the highly purified material from the Sephacryl S-300 chromatography as analyzed on 10% SDS PAGE. Parallel cleavage reactions with Factor Xa were assayed immediately (as described below) or stopped by the addition of 10% trichloroacetic acid and fractionated on 10% SDS PAGE and treated with Coomassie Blue stain or processed for Western blot analysis (data not shown).

Panel A) Fractionation of cell extracts and subfractions of Lys158 and Glu158 human FMO3 MBP fusion proteins analyzed with a 10% SDS PAGE. Panel B) Fractionation of highly purified Lys158 human FMO3 MBP obtained from Sephacryl S-300 chromatography and analysis of chromatography samples on 10% SDS PAGE after treatment with Factor Xa.
Panel A) Lane 1, molecular weight standards;Lane 2, sample of Lys158 human FMO3 MBP from the bacterial lysate; Lane 3, bacterial extract of Lys158 human FMO3 MBP purified with amylose resin chromatography; Lane 4, the pellet from precipitation of amylose resin chromatography purified Lys158human FMO3 MBPwith 7% PEG 8000; Lane 5, the pellet from precipitation of amylose resin chromatography purified Lys158 human FMO3 MBP with 14% PEG 8000;Lane 6, a sample of Glu158 human FMO3 MBP from the bacterial lysate; Lane 7, sample of bacterial lysate of Glu158 human FMO3 MBP purified with amylose resin chromatography; Lane 8, the pellet from precipitation of amylose resin chromatography purified Glu158 human FMO3 MB with 7% PEG 8000;Lane 9, the pellet from precipitation of amylose resin chromatography purified Glu158 human FMO3 MBP with 14% PEG 8000. Panel B) Lane 1, molecular weight standards; Lanes 2 and 3 were column samples from the chromatography and did not contain any detectable protein.Lane 4 showed a significant amount of the enzyme product, that Factor Xa (lower band) cleaved Lys158 human FMO3 MBP (100 kDa) into the native protein of 59 kDA and that the chromatography resolved the human FMO3 from the fusion protein.
Selective Functional Assays for Human FMO3-MBP Activity.
Assay and analysis of human FMO3 MBP N-oxygenation activity with 5-DPT was done as described previously (5, 9, 30). MTSS-oxygenation assays were carried out under the same buffer, temperature, time, and volume conditions as that for theN-oxygenation assays described in the reference cited above. The reaction was stopped by the addition of 700 μl of ice-cold CH3CN/hexane, 80:20, v:v). After the addition of approximately 20 mg NaCl to each incubation, each sample was mixed vigorously for 10 sec. The reactions were then centrifuged at 2,000g for 10 min to separate the organic phase from the aqueous phase. The organic layer was removed and used directly for HPLC analysis. The recovery of MTS and MTSO was judged to be ≥ 90%.
The MTSO product was separated from the substrate MTS with an analytical reversed phase HPLC column (4.6 × 250 mm, Microsorb-MV, Rainin Instruments Co., Inc., Woburn, MA). The HPLC instrument employed was the same as described above for theN-oxygenation reactions. The HPLC conditions for elution were CH3CN/H2O (1:1; v:v) with a gradient flow rate as follows: 1.5 ml/min from 0 to 4 min, 1.5–2.5 ml/min from 4 to 5 min, and 2.5 ml/min from 5 to 15 min. MTSO eluted during the first gradient phase while the substrate eluted during the last phase and were detected at 236 nm with retention times of 2.5 min and 9.5 min, respectively.
Detergent Dependence of Human FMO3-MBP Activity.
After purification of the human FMO3 MBP by amylose resin affinity chromatography, the amount of detergent present was analyzed by extraction of the protein with organic solvents and HPLC analysis of the extracts. This was done for the human glu158FMO3 MBP enzyme although the lys158 enzyme was anticipated to be similar. Human glu158 FMO3 MBP (185 μg, 1.8 nmol) was placed in 0.25 ml buffer (potassium phosphate, pH 8.5) and extracted with 1 ml acetonitrile. The organic fraction was mixed thoroughly, separated by centrifugation, evaporated to dryness, and taken up in methanol for analysis by HPLC. HPLC was done with a Hitachi HPLC system as described above using a C-18 Microsorb reversed phase column (Rainin, Emeryville, CA). An eluent of CH3CN/H2O/CH3OH/NH4OH (62:35:3:0.4; v:v) was used to separate Triton X-100 and other detergents (retention volume 10 ml) from other polar materials. Using a standard curve constructed with between 254 to 5088 pmol of Triton X-100, it was established by HPLC analysis that 0.72 nmol of detergent was present per nmol of enzyme after the amylose resin affinity chromatography step. Enzyme preparations with this negligible amount of detergent was designated “detergent free” human FMO3 MBP. The requirement for additional detergent for optimal activity of the “detergent free” human FMO3 MBP was examined by studying the effect of added Triton X-100 on 5-DPT N-oxygenation. The 5-DPTN-oxygenation enzyme activity assay was done essentially as described above with the exception that a small amount of Triton X-100 (i.e. varied from 0–0.5% detergent as shown in fig.2) was added to the incubation before addition of substrate.

Shows the graphical representation of the specific activity of 5-DPTNO formation in the presence of Glu158 (•) and Lys158 (▴) human FMO3 MBP plotted as a function of the percentage of Triton X-100 detergent added to each incubation.
For the Glu158-human FMO3 MBP, 146.8 μg and for the Lys158-human FMO3 MBP, 200 μg was used in 50 mM potassium phosphate buffer (pH 8.4) containing varying amounts of Triton X-100.
Results
Expression and Purification of Human FMO3 MBP
The PCR products that were designed to give the full length open reading frame cDNA of FMO3-MBP proteins were inserted into a pMAL-c2 expression vector. Endonuclease restriction enzyme analysis and complete DNA sequence analysis (i.e. both strands) of the pMAL-HFMO3 cDNA sequence confirmed that the entire human FMO3 MBP cDNA coding strand was successfully extended and correctly inserted into the expression vector.3
The expression of the pMAL-c2 derived human FMO3 MBP fusion products in the E. coli host bacteria JM109 resulted in production of full length human FMO3 MBP enzymes with a molecular weight of approximately 100 kDa. In addition, several other different sized proteins were retained on the amylose affinity resin (fig. 1). The sizes of the protein products that selectively eluted from the affinity chromatography column ranged from approximately 100 kDa (i.e., the expected size of the full length fusion product) to approximately 43 kD. The 100 kDa protein product was present only in cells that contained the fusion vector and were not observed in cells without a vector or cells transfected only with the pMAL-c2 vector alone (i.e. the pMAL-c2 vector that produced maltose binding protein exclusively as the only amylose binding protein). A common property of the human FMO3 MBPs that selectively bound to the amylose resin was that they were released by maltose. It was likely that the other selectively eluted proteins were the result of expression from the human FMO3 MBP vector, or else contained a maltose binding protein domain and possibly arose either through proteolytic breakdown of the full length protein or were the result of incomplete translation (fig.1A). The results were consistent with the latter hypothesis because the SDS-PAGE pattern of amylose binding proteins observed after expression experiments and the relative intensity of the bands was almost always identical from preparation to preparation, and this implied an inherent stability of the products in the cells or cell extracts employed.
Although bacterial cell lysates derived from JM109 containing pMAL-HFMO3 were active, there was a dramatic increase in specific activity for the conversion of 5-DPT to 5-DPT N-oxide after purifying human FMO3-MBP by amylose affinity chromatography and selective polyethylene glycol (PEG) 8000 precipitation (table1). Further fractionation of the protein on the basis of size showed that the bulk of the human FMO3N- and S-oxygenation activity was associated with the full length product, although a 60 kDa protein also co-eluted with the full length protein. Based on the behavior of the human FMO3 MBP protein on Sephacryl S-300 chromatography, the active fraction eluted earlier, and apparently as a much larger species, than expected for a 100 kDa protein. This result suggested that the active form of the enzyme may be present in higher ordered complexes.
Selective Fractionation of Human Flavin-Containing Monooxygenase Maltose Binding Protein
As shown in fig. 1B, Factor Xa cleaved highly purified human FMO3 MBP to the native 59 kDa FMO3 protein. However, the cleavage was sluggish and during the period required for proteolysis, some degradation of the 59 kDa species was also observed. In a separate, larger scale experiment, Western blot analysis clearly revealed over 80% cleavage of the fusion protein after 3 hr in the presence of Factor Xa. To compare the N-oxygenase activity of the human FMO3 MBP with that of the largely cleaved protein, a preparation of 60–70% cleaved human glu158 FMO3 MBP (i.e. prepared by the action of Factor Xa) and a split sample of non-proteolyzed fusion protein incubated in the presence of NADPH for 2 hr at 37°C were evaluated for 5-DPT N-oxide formation. These two preparations were compared with the activity of a sample of fusion protein stored for 2 hr at 4°C. The 60–70% cleaved protein and the intact fusion protein sample N-oxygenated 5-DPT with essentially equal activity (i.e. 6.25versus 5.57 nmol of 5-DPT N-oxide/min/mg of protein, respectively). The activity of the human FMO3 MBP enzyme incubated in the absence of Factor Xa and the presence of NADPH for 2 hr at 37°C compared favorably with that of enzyme stored at 4°C and then assayed (i.e. 6.27 and 6.73 nmol of 5-DPTN-oxide/min/mg of protein, respectively).
It was notable that inclusion of NADPH during the Factor Xa cleavage reaction markedly stabilized the protein to proteolysis. Thus, in the absence of NADPH, Factor Xa cleavage of human FMO3 MBP gave only 24% of the 5-DPT N-oxygenase activity as that observed in the presence of NADPH. It should be pointed out that Factor Xa-cleaved human glu158 FMO3 possessed many of the unattractive features of highly purified FMO including intractable solubility and other instabilities reported previously (1, 32, 33). In addition to greatly aiding in the purification of human FMO3 MBP enzymes, the attachment of the maltose binding protein domain to the amino terminus of human FMO3 enzymes clearly stabilized the enzyme to thermal inactivation when compared with enzyme activity present in human liver microsomes or produced as a non-fusion protein in E. coli (5-10, 30). Thus, under similar incubation conditions as described above (i.e. in the presence or absence of NADPH), adult human liver microsomes or human FMO3 non-fusion protein lost 100% of 5-DPT N-oxygenating activity in 1 hr at 37°C while in the presence of NADPH, human FMO3 MBP retained greater than 93% of 5-DPT N-oxygenation activity after 2 hr at 37°C.
Physical Chemical and Kinetic Properties of Human FMO3-MBPs.
The regioselective N- and S-oxygenation of a tertiary amine and sulfide, respectively, were examined with lys158 and glu158 of human FMO3 MBPs to investigate selected physical chemical and kinetic properties of these enzymes. Preliminary studies of cDNA-expressed FMO3 MBP enzymes showed that lysates of E. coli cells transformed with pMAL-HFMO3 possessed significant NADPH-dependent 5-DPTN-oxygenation and MTS S-oxygenation activity. Bacterial lysates transformed with vectors that did not contain human FMO3 cDNA did not show significant N- orS-oxygenation activity. Because partial purification of the human FMO3 MBP enzymes by amylose affinity chromatography was fast and efficient and because the fusion protein was equally active as the human FMO3 generated by Factor Xa cleavage, the affinity purified material was used directly in subsequent kinetic experiments. The lys158 and glu158 enzyme preparations were judged to be virtually identical on the basis of SDS-PAGE, Coomassie Blue staining, and Western blot analysis.
Studies showed that cDNA-expressed human FMO3 MBP proteins supplemented with NADPH catalyzed N- or S-oxygenation of 5-DTP and MTS, respectively. For the substrates examined, formation of theS- or N-oxides were a linear function of affinity-purified lys158 and glu158 FMO3 MBP protein concentration (0–100 μg of protein) and of time (0–20 min). Therefore, 30–45 μg of protein and 15 min incubation time was generally used in the kinetic studies described below.
5-DPT N-oxygenation activity of both lys158 and glu158 human FMO3 MBPs were dependent on the pH of the incubation mixture. Both enzymes gave virtually identical pH-rate profiles. 5-DPTN-oxygenation of both lys158 and glu158 human FMO3 MBPs were almost non-detectable below pH 6.5, and the activity rose gradually with a marked increase from pH 7.0 to 8.5. In agreement with previous observations, the optimum pH for 5-DPT N-oxygenation was 10.5, but this probably reflected a significant contribution from the extent of ionization of the tertiary amine substrate (9). The pH generally used for incubations with 5-DTP was pH 8.5 or 9.0. Likewise, MTSS-oxygenation by lys158 and glu158 enzymes of human FMO3 MBP was also dependent on pH. Both enzymes gave similar pH-rate profiles.S-Oxygenation activity rose rapidly between a pH value of 7.0 to 8.0 and achieved a maximal value between pH 9.0 and 9.5. In agreement with previous studies, the true pH optimum of the enzyme is undoubtedly about pH 9.0 (9).
The maximal 5-DPT N-oxygenation activity of both enzymes was observed in the presence of 0.015% Triton X-100 : incubation volume (v:v) (fig. 2). While the preparation of these enzymes included an extensive detergent-free wash of the protein, human FMO3 MBP was avidly bound to the affinity matrix, and a small amount of detergent (i.e. 0.72 nmol per nmol of enzyme as determined by HPLC) remained associated with the protein upon elution from the amylose resin. The amount of detergent that remained with detergent-free enzyme was suboptimal for maximal activity. As shown in fig. 2, an increase of added detergent beyond 0.05% had a negative effect on enzyme activity, especially above 0.5% Triton X-100. Interestingly, the concentrations of Triton X-100 found to be optimal for human FMO3 MBP activity (i.e. 0.015% Triton X-100) were far lower than the amount of detergent that has been commonly used (i.e. 1% or more) for most preparations of FMO enzymes analyzed prior to this study (30-33).
Kinetic Parameters for N-Oxygenation of 5-DPT by Human Lys158 and Glu158-FMO3 MBP
Kinetic constants for 5-DPT N-oxygenation in the presence of human lys158 or glu158-FMO3 MBP were calculated from the rate ofN-oxide formation at variable substrate concentration by the HPLC methods described above. The enzyme of each kinetic determination was judged to be virtually identical on the basis of SDS-PAGE, Coomassie Blue staining, and Western blot analysis. TheKm and Vmaxvalues obtained from double reciprocal plots of velocityversus substrate concentration (i.e.Lineweaver-Burke analysis) were listed in table2. The r values for glu158 and lys158 human FMO3 MBP were 0.99 and 0.98, respectively. The glu158 human FMO3 MBP enzyme displayed a much greater maximal velocity for the oxygenation of 5-DPT than that of the wild type lys158 enzyme. Human glu158 FMO3 MBP showed a slightly higherKm for 5-DPT N-oxygenation than the human lys158 FMO3 MBP variant, but this difference may not be significant. Depending on the enzyme preparation, some intersample variation in the activities for both forms of the enzyme have been observed, but the kinetic differences shown in table 2were much greater than can be explained by any intersample effect.
Kinetic constants for N-oxygenation of 5-DTP catalyzed by the glu158- and lys158-human flavin-containing monooxygenase (form 3) maltose binding fusion proteins2-a
Comparison of Human FMO3 MBP N- andS-Oxygenase Activities with 5-DPT and MTS
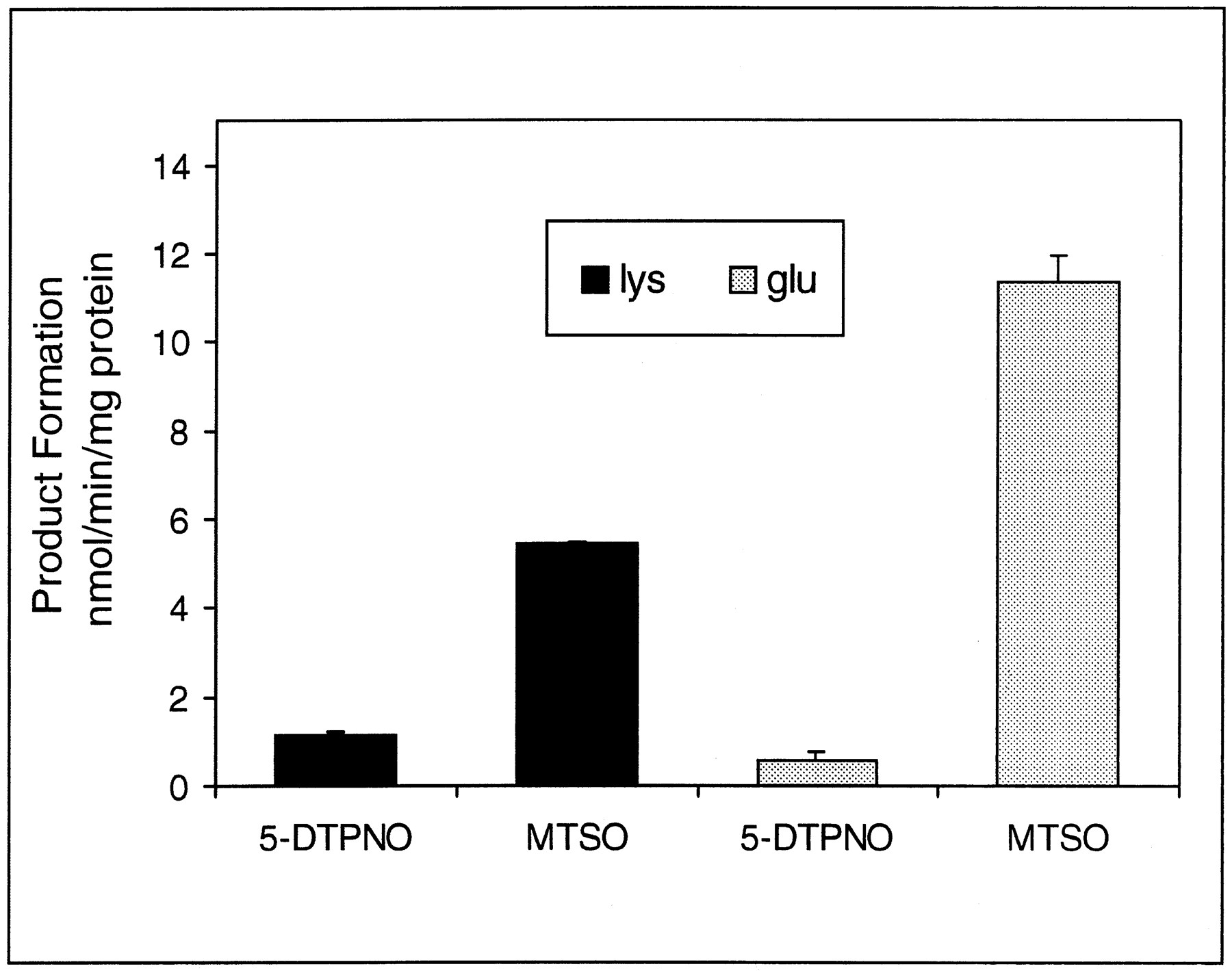
Both human glu158- and lys158 -FMO3 MBP enzymes were examined for 5-DPT tertiary amine N-oxygenation and MTS sulfideS-oxygenation in parallel to unambiguously compare the relative activities between the two enzymes (fig.3). The parallel kinetic analysis was done to avoid any possible problems associated with differences that might arise from kinetic variations resulting from minor enzyme preparation effects. The rate of 5-DPT N-oxygenation and MTSS-oxygenation by human glu158 FMO3 MBP showed that the enzyme was able to discriminate between the tertiary amine and the sulfide substrates examined to a much greater degree than human lys158 FMO3 MBP.

Bar graphs of the relative rate of 5-DPTNO and MTSO formation in the presence of Lys158 (solid black bars) and Glu158 (gray bars) human FMO3 MBP.
For the Lys158 and Glu158variants, 1.13 ± 0.11 and 0.58 ± 0.17 nmol 5-DPTNO/min/mg of protein, respectively, and 5.43 ± 0.04 and 11.37 ± 0.59 nmol MTSO/min/mg of protein, respectively, were formed. Products were determined by the HPLC procedure described in Materials and Methods in the presence of 34 and 51 μg of the Lys158 and Glu158 human FMO3 MBP, respectively.
Conclusion
The maltose binding domain fusion system is an efficient expression system for the production of stable and highly active human FMO3. The MBP fusion allows for an efficient one-step purification procedure that yields enzyme in a substantially purified and highly active state. The utility of human FMO3 fusion proteins has been noted before (1), but this is the first report showing quantification of that proposal. It is notable that the fusion of the MBP domain to human FMO3 markedly decreases the requirement for detergent while at the same time affording a soluble enzyme and actually enhancing the activity and stability of the protein. This is important because essentially detergent-free, soluble human FMO3 MBP can be further purified efficiently with additional steps of fractionation, amylose affinity chromatography, selective PEG 8000 precipitation including size exclusion chromatography either as the fusion protein or, after Factor Xa cleavage, to obtain the native protein. However, because of the enhanced stability, solubility, and essentially equal enzyme activity, use of the fusion protein possesses significant advantages over the cleaved human FMO3. Another attractive feature of soluble, essentially detergent free human FMO3 MBP in xenobiotic or drug metabolism studies arises from the ease of sample preparation and analysis by the types of HPLC procedures routinely used in the presence of essentially detergent-free enzyme. Because most substrates of human FMO3 are lipophilic and often obscured by detergents analyzed in the reversed phase HPLC analytical mode (i.e. or other common means of evaluating substrate oxygenation), a detergent-free enzyme preparation possesses significant advantages over human FMO3 preparations requiring significant quantities of detergent for activity.
The human FMO3 MBP expression system is also adaptable to the expression of other enzymes. We have shown that wild type and variant human FMO3 MBPs can be expressed and purified in active form and studied in detail because of the advantageous properties of the high expression system and the stability of the soluble human FMO3 MBP that are not confounded by membrane-association phenomena. The two human FMO3 enzymes, differing at amino acid position 158, were analyzed with respect to their ability to oxygenate a tertiary amine and a sulfide-containing substrate. Human FMO3 MBPs containing lys158 or glu158 were active against both substrates although the extent of substrate oxygenation showed considerable sensitivity to the particular enzyme used. With the exception of the minimal requirement for detergent and the stabilization by NADPH to thermal inactivation, the physical chemical properties of the human FMO3 MBPs examined (i.e. pH dependence, etc.) were virtually identical to FMO3 expressed inE. coli as a non-fusion protein (5, 30). The stabilization of human FMO3 MBP to proteolysis and thermal inactivation in the presence of NADPH is a notable feature of the fusion protein. It is possible that the NADPH binding domain and substrate binding channel are proximal to the amino terminus of the enzyme and to attachment of the MBP to the amino terminus and protects human FMO3 from degradation.
Both human lys158- and glu158-FMO3 MBP N-oxygenated 5-DPT andS-oxygenated MTS relatively efficiently. However, parallel assays against both substrates revealed that human lys158- and glu158-FMO3 MBP had different capabilities to discriminate between the two substrates (fig. 3). For example, the glu158 enzyme has a much greater relative activity toward the sulfide substrate, MTS (20-fold), than to the tertiary amine substrate, 5-DPT. In contrast, the human lys158 FMO3 MBP enzyme is only about 4.8-fold more efficient at S-oxygenating MTS thanN-oxygenating 5-DPT. While the preliminary results for an admittedly limited number of substrates reported herein strongly indicates that the glu158 enzyme is a distinctive FMO catalyst compared with the wild type lys158enzyme, a more comprehensive description of the difference between these two enzymes is warranted. In this regard, it is notable that the human lys158 FMO3 enzyme reported previously exhibited significant regio- and stereoselective N- andS-oxygenation (5, 30), and this point must be examined with the glu158 enzyme. It is possible that the postulated features of the human FMO3 substrate binding channel may have to be revised in view of the apparently distinctive properties of the enzyme activity of the human glu158 FMO3 enzyme observed in this study.
Because a certain fraction of the human population is homozygous for the gene encoding the glu158 FMO3 enzyme, it is likely that some individuals will be more efficient at oxygenating some nucleophilic heteroatom-containing xenobiotics or drugs than individuals who do not possess glutamic acid at amino acid position 158 of human FMO3. This may have consequences for drug development programs and human drug-drug interactions or other metabolic idiosyncrasies (27).
It is unknown how the naturally occurring human FMO3 enzyme polymorphism (i.e. lys158 and glu158 FMO3) affects the metabolism of xenobiotics that humans are exposed. For example, although it has been reported that human FMO3 is the enzyme responsible for conversion of TMA to its deodorification product TMANO, it is unknown what role the human FMO3 enzymes reported herein play in TMA tertiary amineN-oxidative metabolism in vivo. Likewise, it is unknown what role human FMO3 enzymes play in modulating the metabolism of other endogenous or xenobiotic amines that may be associated with some disease condition. It is possible that variation at the FMO3 locus may contribute in certain populations to disease associated with environmental exposure to procarcinogens. Further studies of the role of human FMO3 enzymes in the metabolism of exogenous and endogenous amines may lead to a clearer understanding of the role of human FMO3 in xenobiotic detoxication and human disease.
Acknowledgments
The authors are grateful to our collaborators who supplied some of the biological agents necessary for some of the above studies: Professor Alan Rettie (University of Washington) for anti-monkey liver FMO antibody, Dr. Randall Howard (Seattle Biomedical Research Institute) for the pMAL vector and Dr. K. Kujikawa and Professor E. Davie (University of Washington) for Factor Xa. We acknowledge the stimulating conversations with Drs. Eileen Treacy (McGill University, Montreal, Canada) and Susan Forrest (Royal Children’s Hospital, Melbourne, Australia) concerning trimethylaminuria patients.
Footnotes
-
Send reprint requests to: Dr. John Cashman, Seattle Biomedical Research Institute, 4 Nickerson Street, Suite 200, Seattle, WA 98109-1651.
-
This work was financially supported by a grant from NIH (GM 36426)
-
↵2 Restriction length polymorphism and oligonucleotide sequencing studies showed codon 158 encoded either amino acids glu or lys at approximately equal allele frequencies for the caucasian populations examined: E. Treacy, R. Youil, S. Forest, and M. Knight, unpublished data.
-
↵3 The revised full length human FMO3 cDNA sequence was recently reported: Proc. Natl. Acad. Sci. USA 92,9910 (1995) and Eur. J. Biochem. 235, 683 (1996).
- Abbreviations used are::
- TMA
- trimethylamine
- TMANO
- trimethylamine N-oxide
- 5-DPT
- 10-(N,N-dimethylaminopentyl)-2-(trifluoromethyl)phenothiazine
- 5-DPTNO
- 10-(N,N-dimethylaminopentyl)-2-(trifluoromethyl)phenothiazineN-oxide
- MTS
- methyl p-tolyl sulfide
- MTSO
- methyl p-tolyl sulfoxide
- HFMO3
- human flavin-containing monooxygenase (form 3)
- IPTG
- isopropyl-β-D-thiogalactopyranoside
- PMSF
- phenylmethylsulfonylfluoride
- DETAPAC
- diethylenetriaminepentaacetic acid
- MBP
- maltose binding protein
- PCR
- polymerase chain reaction
- HPLC
- high performance liquid chromatography
- DCC
- dicyclohexylcarbodiimide
- THF
- tetrahydrofuran
- TLC
- thin layer chromatography
- SDS PAGE
- sodium dodecylsulfate polyacrylamide gel electrophoresis
- PEG 8000
- polyethylene glycol 8000
- Received December 23, 1996.
- Accepted March 31, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}