


Application of an In Vitro Glucuronosyltransferase Assay
Abstract
An in vitro glucuronidation assay was used to optimize a series of N-hydroxyurea-containing 5-lipoxygenase inhibitors for metabolic stability. The glucuronidation of these compounds in cynomolgus monkey microsomes followed Michaelis-Menten kinetics allowing calculation of Vmax andKM . The Vmax values ranged from 0.02 to 7.9 nmol/min/mg microsomal protein, a 400-fold difference, whereas KM ranged from 204 to 2500 μM, only a 12-fold difference. In vitro intrinsic clearance values (CLint) were calculated for 18 compounds tested in the kinetic assay and compared with the in vivo plasma clearance (CLp ) calculated from intravenous studies done in cynomolgus monkeys. These initial results suggested a relationship between the in vitro CLint and in vivo duration as defined byCLp . A more rapid in vitro assay was developed in a 96-well format using a single concentration of substrate (100 μM) from which a glucuronidation rate was calculated. The results from this assay for 40 compounds correlated with in vivo plasma clearance (r = 0.57). This more efficient assay was used to test more than 100 compounds and develop structure–metabolism relationships based on metabolic stability and improved duration. The culmination of this effort contributed to the discovery of ABT-761, a 5-lipoxygenase inhibitor with in vivo duration in monkey improved 40-fold over the first generation inhibitor. Further studies performed in human liver microsomes demonstrated a similar trend that was corroborated by the 8-fold increase in duration after oral dosing in humans observed with ABT-761.
Leukotrienes have been implicated as inflammatory mediators in diseases such as asthma and rheumatoid arthritis. Leukotriene biosynthesis is initiated by the enzyme 5-LO.1 The inhibition of 5-LO activity has been a therapeutic target for a number of years (1) and has resulted in the discovery of zileuton (Zyflo), a potent, orally bioavailable inhibitor, currently in clinical trials to treat asthma (2). Zileuton contains an N-hydroxyurea and a single chiral center. It is administered as a racemic mixture of the R(+)- and S(−)-enantiomers. The major route of metabolism for zileuton in humans and cynomolgus monkeys has been reported to be glucuronidation at the N-hydroxyurea substituent with subsequent urinary excretion (3). The masking of theN-hydroxy moiety has been shown to result in compounds lacking 5-LO potency (4); therefore, the formation of a glucuronide at this position would result in pharmacological inactivation. The half-life of zileuton in humans has been shown to be 2.4 hr after oral dosing and more rapid in monkey (t1/2 = 0.4 hr) (5), and the ex vivo efficacy of this compound has been shown in several species to follow the plasma levels of the parent compound closely (6). Efforts to increase the in vivoduration of this chemical series has involved chemical modifications that would decrease this rate of metabolism, yet maintain theN-hydroxyurea pharmacophore necessary for 5-LO inhibitory potency (7).
The testing of a large number of compounds in vivo can be time-consuming, tedious, and costly, especially when the species of interest is a nonhuman primate. In recent years, using in vitro metabolism methods has become a useful way to screen large numbers of test compounds rapidly with respect to a specific metabolic reaction (8-10). Determinations of CLint with regard to a single enzyme reaction in vitro can be shown to correlate to in vivo CLh by mathematical derivations and can be shown to predict in vivoduration through subsequent intravenous dosing studies with selected compounds.
To assist in the effort to identifyN-hydroxyurea–containing compounds with improved duration, an in vitro UDPGT assay was developed using liver microsomes from cynomolgus monkeys. The following study will describe how three structural areas of the N-hydroxyurea compounds were modified and how these modifications affected the in vitroUDPGT rate and the in vivo duration. The UDPGT assay was used to develop structure–activity relationships that led to compounds with increased duration in cynomolgus monkeys. Glucuronidation rates for selected compounds were also obtained from human liver microsomes that compared closely to the rates from monkey microsomes. The results of these studies led to the discovery of a compound (ABT-761) with a 16-hr half-life in humans.
Materials and Methods
Chemicals.
Protein concentrations were determined with the Pierce BCA protein kit (Rockford IL). Acetohydroxamic acid and triethylamine were also purchased from Pierce. UDPGA, tetrabutylammonium hydrogen sulfate, potassium chloride, and sodium phosphate were purchase from Sigma Chemical Co. (St. Louis, MO). Methanol, acetonitrile, and tetrahydrofuran were obtained from EM Science (Gibbstown, NJ).
Animals and Tissue Samples.
Cynomolgus monkeys (3–8 kg) were obtained from Charles River Primates (Houston, TX). Cynomolgus monkeys liver samples were obtained from euthanized control animals and were frozen at −80°C. Human liver specimens were obtained from the National Disease Research Institute (Philadelphia, PA).
Enzyme Kinetic Studies.
Frozen liver samples were thawed on ice and microsomes prepared as described previously (7). Protein concentrations were determined using the Pierce BCA protein assay.
Enzyme kinetic studies were performed at 37°C with 1 mg/ml of microsomal protein containing 5 mM MgCl2, 0.1 M Tris (pH 7.4), 0.3% Triton X-100, and test compound in DMSO (final concentration: 1% in assay). Reactions were initiated after a 5-min preincubation with the addition of 3–10 mM UDPGA. The test compound concentration was varied from 50 to 2000 μM and incubation times controlled to keep the substrate turnover <15%. Samples were quenched with 2 volumes of acetonitrile and centrifuged to remove precipitated protein and analyzed by HPLC.
UDPGT Assay.
The UDPGT assay was formated in a 96-well tray using polypropylene tubes that allowed the use of multichannel pipetters and/or robotics for sample handling. Incubations were performed at 37°C as described previously (7).
HPLC.
Samples from the in vitro assays were analyzed by reversed-phase HPLC using a Waters WISP autosampler, an LKB model 2150/2152 pump, a Laboratory Data Control variable wavelength UV detector, and a Spectra-Physics 4200 Integrator. Little Champ C18 columns (Regis Chemical, Morton Grove, IL) were used with mobile phases containing 0.5 mM acetohydroxamic acid with 5–12.5 mM tetrabutylammonium hydrogen sulfate and 10–25 mM potassium phosphate at pH’s ranging from 3 to 6.5, with varying amounts of acetonitrile, methanol, and/or tetrahydrofuran. The flow rates were 0.4–0.5 ml/min, and peaks were monitored by measuring UV absorbance at 230 or 260 nm.
To determine plasma levels for duration studies, plasma samples were thawed, 2 volumes of methanol were added, and precipitated proteins were removed by centrifugation. Supernatants were injected directly onto a C18 reversed-phase Adsorbosphere HL-7 μm column (Alltech Associates, Deerfield, IL) or onto a Little Champ and eluted with mobile phases consisting of 5 mM acetohydroxamic acid with 8 mM triethylamine acetate at pH 4 with varying amounts of acetonitrile, methanol, and/or tetrahydrofuran. The flow rates were 0.4–1.0 ml/min, and peaks were monitored by measuring UV absorbance at 230 or 260 nm and quantified using an external calibration curve. Pharmacokinetic parameters were calculated using NONLIN.
In Vitro Kinetics.
Velocities were calculated based on appearance of conjugates over time.Vmax and KM
were calculated from linear regression analysis of Lineweaver-Burk and Eadie-Hofstee plots using KaleidaGraph software (Synergy Software, Reading, PA). In vitro clearance values were calculated using (Vmax/KM
) as derived from the Henri-Michaelis-Menten equation
In Vivo Studies.
Compounds for intravenous dosing were dissolved in DMSO and formulated with ethanol, PEG-400 or TRAPSOL (CTD, Inc., Alachua, FL), and saline. They were administered to cynomolgus monkeys in dose volumes from 1 to 2 ml/kg via the saphenous vein. Heparinized blood samples were withdrawn at various times after dosing from the femoral vein. Samples were centrifuged, the plasma was removed, and frozen at −20°C until assayed.
In Vivo Kinetics.
CLp
was calculated using the elimination rate constant of the terminal phase (min) multiplied by theVd
(ml/kg) as shown in the equation below.
Results
As an aid to developing structure–activity relationships, theN-hydroxyurea compounds were divided into three areas for structural modification: the template, the linking group, and the pharmacophore (fig. 1). Although it is the site of metabolism, previous work had already defined the pharmacophore in zileuton as optimal for potency and selectivity; therefore, it was not modified in the search for metabolically stable compounds. The template and link components were the major focus of medicinal chemistry efforts to optimize in vivo duration in the cynomolgus monkey (13).

Structure of zileuton showing the three groups used to define the structure–metabolism relationships of the N-hydroxyureas: template, link, and pharmacophore.
Glucuronidation is the major route of metabolism in monkeys and humans.
Glucuronidation of zileuton in cynomolgus monkey liver microsomes followed Michaelis-Menten kinetics over a 50–2000 μM concentration range (data not shown). The removal of a methyl group from the linkage group resulted in an increase in the Vmax as shown in table 1 and resulted in a 6-fold increase in the in vitro CLint. The half-life of the methylene analog was 2-fold shorter than zileuton when dosed intravenously in monkeys. Higher clearances for the methylene analogs were noted, with two other sets of compounds demonstrating a correlation between in vitro CLint and in vivo duration (table 1).
In vitro clearance and in vivo duration of methyl branch and methylene analogs
Structure–Activity Relationships Using the UDPGT Assay.
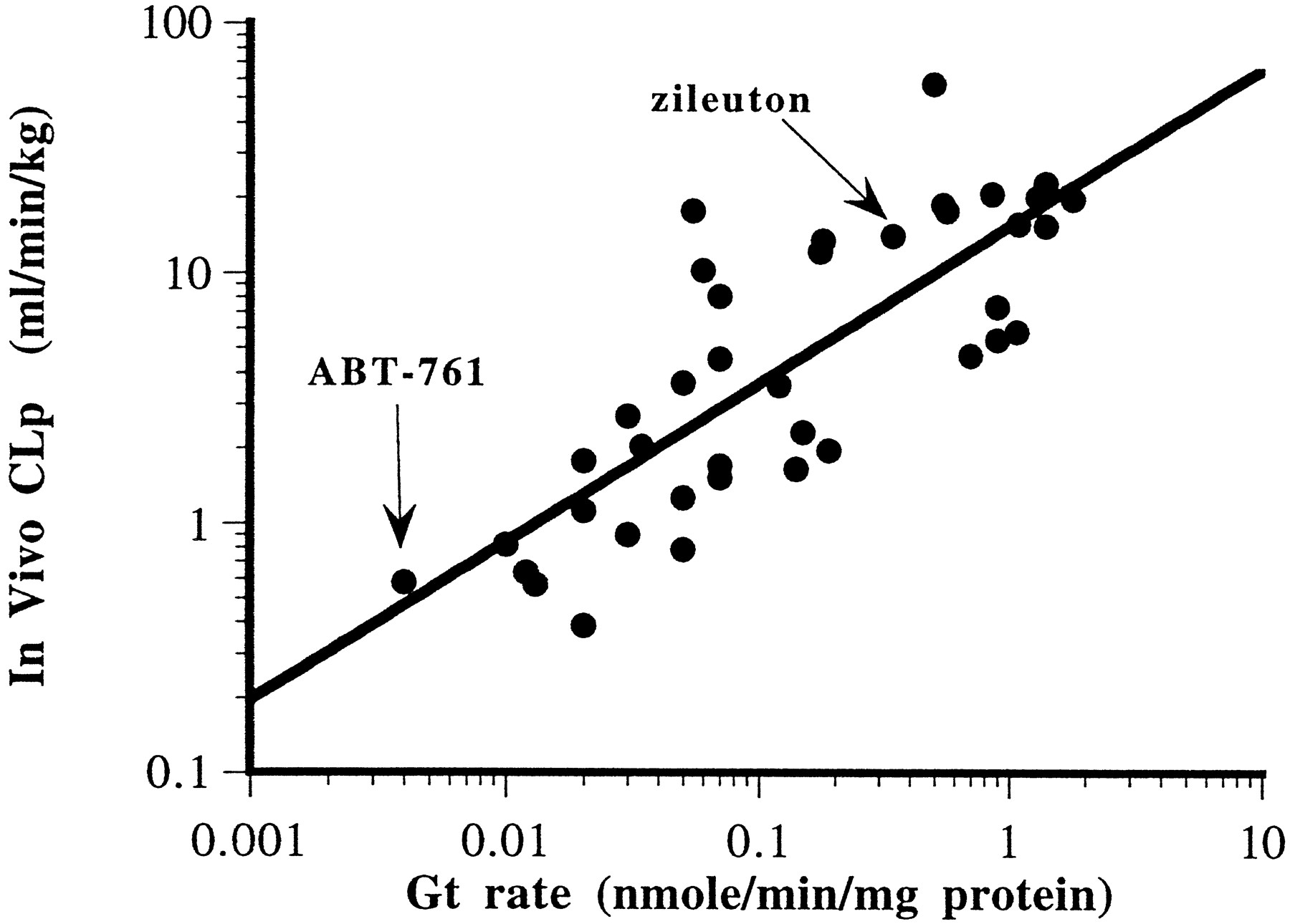
The UDPGT assay format was developed to facilitate the rapid testing of a number of structural analogs, up to 15 in one assay, whereas minimizing the amount of microsomal protein used. The 96-well array made use of automated 8- or 12-channel pipettors and/or robotics possible. The test compound concentration was 100 μM, less than theKM for the 18 compounds analyzed kinetically. A reference standard (the methylene analog of zileuton) was included in each assay, and results were normalized to that standard rate. Comparisons were made between the UDPGT rate and in vivo CLp (fig. 2) and a correlation determined (r = 0.57). The measuredCLp values were less than the hepatic blood flow that combined with the knowledge that glucuronidation is a major route of metabolism for these compounds suggested that clearance was primarily through the liver. Structure–activity relationships were developed further using the more rapid UDPGT assay.

Relationship between UDPGT rate and in vivo CLp for 40 compounds.
UDPGT rates were determined in cynomolgus monkey microsomes based on appearance of glucuronide conjugate over time at a single (100 μM) substrate concentration. In vivo CLp was determined for each compound from kel andVd data obtained during intravenous dosing studies in cynomolgus monkey (n = 1 in most cases). Intravenous doses varied from 20 mg/kg to 1 mg/kg. r = 0.57.
Selection of the Linkage Group.
In addition to the methyl branch linkage group, several other linkage moieties were investigated. Because the template had yet to be optimized, compounds with several different templates were used to test each linkage group and the data collated. The results are shown in table 2 where a trend could be established. Rapid UDPGT rates and short in vivo half-lives were consistently observed with compounds containing the cyclopropyl linkage. Similarly, rapid UDPGT rates were observed with compounds containing the cis double bond configuration; however, compounds having the trans configuration demonstrated markedly lower UDPGT rates and longer in vivo half-lives. Compounds with the acetylene linkage group were generally found to have lower UDPGT rates than any of the links tested. This lower UDPGT rate resulted in the longest in vivo duration in monkey.
In vitro glucuronidation rates and in vivo duration of different linkage groups
Selection of the Template.
Many variations of the template group were tested in the UDPGT assay. It was clear that other forms of metabolism could affect this portion of the molecule, and every effort was made to block obvious sites for hydroxylation, demethylation, and N-deacylation. Compounds containing the benzthiophene template found in zileuton demonstrated more rapid rates of glucuronidation than compounds with other templates (table 3). Conversely, the use of simple furan or thiophene ring templates could abolish UDPGT activity completely for otherwise identical compounds. Biaryl templates such as phenoxyphenyl, phenoxyfuran, and phenylthiophene also demonstrated reduced rates of glucuronidation. A few examples of the template analogs tested are shown in table 3, comparing their UDPGT rates with their CLp and intravenous half-lives.
In vitro glucuronidation rates and in vivo durations for different template groups
Stereochemistry of the Methyl Branch Linkage Group.
N-Hydroxyurea compounds containing a methyl branch on the linking group had already shown longer in vivo durations than their methylene analogs, which encouraged further work with this methyl branch series. The addition of this methyl group introduced a chiral center and resulted in a compound with separable R- and S-forms; however, these enantiomers exhibited nearly equivalent 5-LO inhibitory activities (14). In contrast, it was found that, when these individual enantiomers were tested in the UDPGT assay, a large difference in the rate of glucuronidation was observed and that this in vitro data compared well with the in vivodurations observed after intravenous dosing. Three examples comparingR- and S-enantiomers are shown in table4. The R-form of compound 17 demonstrated the lowest UDPGT rate yet observed and the longestin vivo duration. This compound was subsequently identified as a clinical candidate and named ABT-761.
In vitro glucuronidation rates and in vivo durations for R- and S-enantiomers of N-hydroxyurea inhibitors in cynomolgus monkey
UDPGT Rates with Human Liver Microsomes.
The glucuronidation of zileuton had been shown to follow Michaelis-Menton kinetics in human liver microsomes (15, 16). Therefore, selected compounds were tested in the UDPGT assay using human microsomes to determine if the trend established with the monkey microsomes would translate to the clinical setting. The duration comparisons between monkeys and humans were made using plasma half-life after oral administration in both species, because intravenous data were not available in humans. Interestingly, the plasma duration in monkeys after oral dosing was very comparable with the intravenous studies, indicating that absorption and/or first-pass gastrointestinal metabolism were not issues with these compounds. As shown in table5, monkey and human microsomal glucuronidation rates of the N-hydroxyurea compounds were quite similar, and the in vitro assay provided a rank order for the in vivo duration in humans.
Comparison of glucuronidation rates from cynomolgus monkey and human liver microsomes with in vivo duration in both species
Discussion
The hepatic microsomal in vitro UDPGT assay described herein reliably ranked the in vivo duration of theN-hydroxyurea compounds in cynomolgus monkey. The initial results showing a correlation between clearances calculated fromin vitro data and in vivo values were encouraging, and stimulated the development of the more rapid UDPGT assay that provided a velocity measurement at a constant concentration of substrate (<KM ). The ability of this assay to test over 100 compounds for metabolic stability yielded the structure–activity relationships necessary to optimize theN-hydroxyurea 5-LO inhibitors for extended in vivo duration.
The use of the UDPGT assay to optimize in vivo duration involved several assumptions. First, that CLp equates with CLh , meaning that the disappearance of compound from the plasma compartment was indeed a reflection ofCLh and that no other elimination (or distribution) processes were occurring. This assumption was supported by the knowledge that glucuronidation was the major route of metabolism for zileuton in the monkey.2However, recently, it has been reported that sulfoxidation and hydroxylation by CYP is a minor route of metabolism for zileuton in humans (17). The second assumption was that the pharmacokinetics were linear or first order in vivo ([S] <KM ) as the kinetics were linear in vitro ([S] < KM ). The 20 mg/kg intravenous doses used in earlier in vivo studies to provide reliable half-life measurements were later replaced with lower 1 mg/kg doses as the in vivo durations increased. This 20-fold difference in the intravenous doses administered could affect both Vd and kel, even if [S] < KM . The in vitrodose [S = 100 μM] was chosen as lower than the lowest KM , but also as a concentration likely to provide measurable glucuronide levels. This in vitroconcentration was 2-fold lower than the lowestKM and 20-fold lower than the highestKM measured—a difference that could result in 10-fold variations in the glucuronidation rates observed. Third, that compound distribution between red blood cells and plasma proteins was a constant ratio for all compounds tested and that the plasma protein binding was constant for all analogs. Distribution between red blood cells and plasma had not been determined for this series of compounds. However, plasma protein binding in cynomolgus monkeys had been determined for a select group of compounds and was reported to be 93% for zileuton (18) and 99% for ABT-761.2 A further assumption was that metabolic switching was not occurring, where a compound would become a less active substrate for the UDPGT enzyme and thereby a more likely substrate for another metabolizing enzyme system. The lack of metabolic switching was, in part, engineered by choosing templates that would be more stable to other routes of metabolism and was demonstrated by relationship observed betweenCLint and CLp .
Data presented in fig. 2 described the correlation observed between theGt rate and CLp for theN-hydroxyurea compounds tested. The Gt assay could provide information to rank order compounds for improved in vivo duration and could indicate which structural modifications would exhibit longer half-lives; however, the correlation coefficient of 0.57 was not remarkable. As discussed in the previous paragraph, differences in the intravenous doses administered, the use of a singlein vitro concentration to determine the Gt rate, dissimilarities in plasma protein binding, and possible metabolic switching were all variables in this approach and could result in significant differences between CLint andCLp , thereby reducing the correlation. For example, six compounds exhibited a Gt rate of ∼0.08 nmol/min/mg protein, yet there was a 10-fold difference in theCLp reported. This discrepancy indicated that it would be difficult to predict accurately the duration of these compounds in monkeys; but, overall, the information provided could help to preselect compounds with low Gt rates for furtherin vivo testing, thereby streamlining the drug discovery process by significantly reducing the number of animal studies needed to identify a clinical candidate.
The potential benefits of in vitro modeling in phase I and phase II metabolizing systems have been described. The increased knowledge of the UDPGT and CYP enzyme systems has combined with the improved methods for cultured hepatocyte preparations, liver tissue slices, and cloning of drug-metabolizing enzymes to enhance greatly the use of this technology in the preclinical setting. Houston (10) collated clearance values from the literature for 25 drugs tested in rat microsomal and hepatocyte preparations for which in vivodata was also available and was able to demonstrate encouraging correlations from data covering four orders of magnitude. Mistry and Houston (9) compared hepatic and intestinal glucuronidation bothin vitro and in vivo for morphine, naloxone, and buprenorphine, and found that a comparative approach of rank ordering to be a more successful means of correlating in vitro andin vivo data than the generation of absolute values of intrinsic clearance. Chiu (19) discussed the use of in vitromodeling to identify species differences in metabolic profiles for losartan in both phase I and phase II systems. The results of thein vitro studies were found to compare well with thein vivo observations in animal experiments and provided a basis for predicting metabolism in humans before clinical testing.
The recent advances in vitro methodologies for the study of drug metabolism combined with the increased availability of human liver tissue have contributed toward reducing the use of animal models and guiding the drug discovery process toward the rational design of longer acting compounds in humans. The structure–metabolism relationships needed to improve compound duration were defined by the UDPGT assay in a matter of weeks and at minimal cost, compared with the months of effort that would have been needed to perform a similar number of in vivo pharmacokinetic studies. Using the UDPGT assay with human liver microsomes helped to identify ABT-761, a second generation 5-LO inhibitor with an 8-fold longer duration in humans than zileuton (20). The success of this current endeavor will provide further impetus for this growing trend in the pharmaceutical industry.
Acknowledgments
The author would like to thank the following personnel of Abbott Laboratories: Ascension Hernandez and John Granger for their expert technical assistance with the monkey studies, and Dr. A David Rodrigues for critically evaluating this manuscript.
Footnotes
-
Send reprint requests to: Jennifer J. Bouska, Department 47K, Building AP9, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, IL 60064.
-
↵2 J. Machinist, personal communication.
- Abbreviations used are::
- 5-LO
- 5-lipoxygenase
- zileuton (ABT-077)
- N-(1-benzo[b]thien-2-ylethyl)-N-hydroxyurea
- t1/2
- plasma half-life
- CLint
- intrinsic clearance [Vmax/KM ]
- CLh
- hepatic clearance
- UDPGT
- UDP glucuronosyltransferase
- ABT-761
- (R)-N-(3-(5-(4-fluorophenylmethyl)-2-thienyl)-1-methyl-2-propynyl)-N-hydroxyurea
- BCA
- bicinchoninic acid
- UDPGA
- UDP glucuronic acid
- DMSO
- dimethylsulfoxide
- Vd
- volume of distribution
- CLp
- plasma clearance
- CYP
- cytochrome P450
- Received January 21, 1997.
- Accepted April 23, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}