


Abstract
In vitro studies were conducted to identify the hepatic cytochrome P450 (CYP) forms involved in the oxidative metabolism of [14C]ABT-761 and itsN-dehydroxylated metabolite, [14C]ABT-438, by human liver microsomes. The two compounds were metabolized by parallel pathways, to form the corresponding methylene bridge hydroxy metabolites. There was no evidence of sulfoxidation and/or ring hydroxylation. Over the ABT-761 and ABT-438 concentration ranges studied (1–300 μM), the rate of NADPH-dependent hydroxylation was linear with respect to substrate concentration ([S]) and did not conform to saturable Michaelis-Menten kinetics. Under these conditions ([S] <KM ), the intrinsic clearance (Vmax/KM ) of ABT-438 was 10-fold higher than that of ABT-761 (1.7 ± 0.8vs. 0.17 ± 0.06 μl/min/mg, mean ± SD,N = 3 livers). The hydroxylation of both compounds was shown to be highly correlated (r = 0.83,p < 0.01, N = 11 different human livers) with CYP3A-selective erythromycin N-demethylase activity, and the correlation between ABT-761 hydroxylation and tolbutamide hydroxylase (CYP2C9-selective) activity (r= 0.63, p < 0.05, N = 10) was also statistically significant. Ketoconazole (2.0 μM), a CYP3A-selective inhibitor, inhibited the hydroxylation of both compounds by 53–67%, and sulfaphenazole (CYP2C9-selective) decreased activity by 10–20%. By comparison, α-naphthoflavone, a known activator of CYP3A, stimulated the hydroxylation of ABT-761 (8-fold) and ABT-438 (4-fold). In addition, the abundance-normalized rates of cDNA-expressed CYP-dependent metabolism indicated that hydroxylation was largely mediated (66–86%) by CYP3A(4). Therefore, it is concluded that the hydroxylation of ABT-761 and ABT-438 (≤10 μM) is primarily mediated by CYP3A, although CYP2C9 may play an ancillary role.
Much attention has been focused on the design of potent and selective 5-lipoxygenase inhibitors, because this enzyme catalyzes the second and third steps of leukotriene A4biosynthesis and leukotrienes are considered pathophysiological mediators of inflammatory diseases (Brooks et al., 1993;Carter et al., 1991). One such example is zileuton (ABT-077) [N-(1-benzo[b]thien-2-ylethyl)-N-hydroxyurea], a substituted hydroxyurea that has potential clinical applications in the treatment of diseases such as rheumatoid arthritis, inflammatory bowel disease, psoriasis, and asthma (Brooks DW et al., 1993; Carter et al., 1991; Weinblatt et al., 1992; Isreal et al., 1990). Like zileuton, ABT-761 [(R)-(+)-N-3-[5-(4-fluorobenzyl)thiophen-2-yl]-1-methyl-2-propynyl-N-hydroxyurea] (fig. 1) is a substituted hydroxyurea derivative and is a potent inhibitor of 5-lipoxygenase (Brooks CDWet al., 1995; Rosenberg et al., 1995). However, ABT-761 exhibits a more favorable pharmacokinetic profile, compared with zileuton (unpublished observations). In addition, unlike zileuton, ABT-761 fails to elicit a clinically relevant drug interaction with theophylline (Wong et al., 1998; Grannemanet al., 1995). This finding suggests that the CYP2 profile of ABT-761 differs from that of zileuton.

Proposed ABT-761 and ABT-438 oxidative pathways.
*, Position of the chiral center and carbon-14 label.
The results of recent in vitro (precision-cut human liver slices) and in vivo (human 14C absorption/distribution/metabolism/excretion) metabolism studies have indicated that ABT-761 is primarily metabolized via directN-hydroxy glucuronidation (Machinist JM, unpublished observations). However, in both cases, the methylene bridge hydroxy metabolite of ABT-761 (ABT-515) was also detected (fig. 1). By analogy with the reduction of zileuton to ABT-193 (Machinist et al., 1995), it appears that unabsorbed ABT-761 is also reduced by gut bacteria, because the products of the metabolism of14C-labeled ABT-438 [(R)-N-3-[5-(4-fluorobenzyl)thiophen-2-yl]-1-methyl-2-propynylurea] have also been observed in human urine and feces. This implies that ABT-438 can be metabolized via CYP-mediated hydroxylation. Therefore, we sought 1) to define the NADPH-dependent metabolism of [14C]ABT-761 and [14C]ABT-438 in a panel of human liver microsomes, 2) to identify the human CYP forms that catalyze the metabolic reactions, and 3) to compare the CYP profile of ABT-761 with that of zileuton.
Materials and Methods
Chemicals.
Unlabeled ABT-761 and ABT-438 and corresponding hydroxy metabolites (e.g. ABT-515), [14C]ABT-761 (152 μCi/mg; radiochemical purity, >96%), and [14C]ABT-438 (159 μCi/mg; radiochemical purity, >96%) were synthesized at Abbott Laboratories. All other, commercially available reagents and solvents were of either analytical or HPLC grade. Microsomes prepared from B lymphoblast cells (AHH-1 TK±) containing cDNA-expressed CYP1A2, CYP2D6-Val374, CYP3A4, CYP2C19, CYP2C9-Arg144, CYP2A6, or CYP2E1 were obtained from Gentest Corp. (Woburn, MA). Microsomes prepared from B lymphoblastoid cells containing the selectable plasmid vector without a cDNA insert, which were essentially devoid of CYP, were also obtained from Gentest. Human liver microsomes were prepared, stored, and characterized as previously described (Machinist et al., 1995; Rodrigues et al., 1994, 1995, 1996; Rodrigues, 1994).
Incubation of ABT-761 and ABT-438 with Native Human Liver Microsomes.
In vitro incubations were performed at 37°C in a Dubnoff shaking water bath, using 12- × 75-mm borosilicate glass disposable culture tubes (Machinist et al., 1995). Briefly, the final assay volume was 0.4–1.0 ml and contained the following: 0.1 M potassium phosphate buffer (pH 7.4), 0.1 mM EDTA, 15 mM magnesium chloride, 4.0 mM NADP+, 10 mM D-glucose-6-phosphate, 2.0 units/ml D-glucose-6-phosphate dehydrogenase (Sigma type VII, from baker’s yeast), 0.5–2.0 mg/ml microsomal protein, and 1–300 μM [14C]ABT-761 or [14C]ABT-438. The reactions were initiated by addition of the NADPH-generating system, after a 3-min preincubation period (37°C, open to air), and were stopped by addition of an equal volume of chilled methanol to precipitate the protein. The samples were centrifuged (2000g, 10 min) and analyzed directly by HPLRC. Under these assay conditions, reactions were linear with respect to protein concentration and time of incubation.
Kinetic Analysis.
The untransformed data were fitted to a one- or two-enzyme model (PCNONLIN, version 4.0; Statistical Consultants, Lexington, KY). However, because of poor drug solubility at higher concentrations (>0.3 mM), it was not possible to obtain apparentKM and Vmaxestimates. CYP was not saturated over the drug concentration ([S]) range studied, with [S] ≪ KM . The relationship between [S] and the initial reaction velocity (v) is described by the Michaelis-Menten equation,i.e. v = (Vmax·[S])/(KM + [S]). For [S] ≪ KM ,v =Vmax/KM ·[S]; intrinsic clearance (Vmax/KM ) is obtained from the slope of a linear plot of v vs.[S].
Measurement of CYP Form-Selective Activities.
Liver microsomal total CYP contents were determined by the method ofOmura and Sato (1964). Erythromycin N-demethylase (final concentration of substrate in incubation medium, 0.5 mM, CYP3A),N,N-dimethylnitrosamine N-demethylase (0.2 mM, CYP2E1), [1,2-3H2]tolbutamide methyl hydroxylase (1.0 mM, CYP2C9/10), COU 7-hydroxylase (0.2 mM, CYP2A6), [O-methyl-14C]dextromethorphanO-demethylase (20 μM, CYP2D6), 7-ethoxyresorufinO-deethylase (2.5 μM, CYP1A2), (S)-(+)-[4-14C]mephenytoin 4′-hydroxylase (0.5 mM, CYP2C19), zileuton/ABT-193 hydroxylase (CYP2C9/CYP1A2), and zileuton/ABT-193 sulfoxidase (CYP3A/CYP2C9) activities were measured as previously described (Machinist et al., 1995; Rodrigues, 1994; Rodrigues et al., 1994,1996). [3H]Terfenadine t-butyl hydroxylase activity was also measured, using HPLRC (Rodrigues et al., 1995). Correlation coefficients were determined graphically using CA-Cricket Graph software (Computer Associates, San Jose, CA).
CYP-Selective Inhibitors.
Inhibition studies were carried out at a final ABT-761 or ABT-438 concentration of 10 μM [at therapeutic doses (150 mg), the maximal plasma concentration of ABT-761 (free and protein-bound) is typically 5.7 μg/ml (18 μM)]. Where possible, mechanism-based inhibitors or relatively high-affinity reversible inhibitors (Ki ≤ 1.0 μM) or cosubstrates (KM ≤ 5.0 μM) were used. In all cases, the final concentration of the inhibitor (cosubstrate) exceeded (≥10-fold) its apparent Ki (orKM ). In addition, the inhibitors were dissolved in water or methanol (final volume, ≤0.5%, v/v).
Incubation with B Lymphoblastoid Cell Microsomes Containing cDNA-Expressed CYP Forms.
Incubations were conducted at 37°C, in 1.5-ml polypropylene centrifuge tubes (final volume, 0.25–0.6 ml), and were carried out as described for liver microsomes (see above). The final concentration of [14C]ABT-761 or [14C]ABT-438 was 10 μM. Samples were preincubated for 5 min, and the reaction was initiated by the addition of 25 μl of rapidly thawed (37°C) microsomal protein (final concentration, 1.0 mg/ml). For control incubations, microsomes prepared from human B lymphoblastoid cells without vectors were used. Incubations were terminated at the specified times with ice-cold methanol, as described previously. The various preparations of cDNA-expressed CYP had been characterized by the manufacturer. All incubations with cDNA-expressed CYP2A6 were carried out with 50 mM Tris-HCl buffer (pH 7.4) containing 0.1 mM EDTA.
For all CYP proteins tested, the reaction rates (in picomoles per hour per picomole of CYP) were normalized (picomoles per hour per picomole · picomole per milligram) with respect to the corresponding nominal specific content of each CYP in native human liver microsomes (Shimadaet al., 1994; Lecoeur et al., 1994). The normalized rates (in picomoles per hour per milligram) were then added, and the normalized rate for each CYP was expressed as a percentage of the total normalized rate. No attempt was made to extrapolate the rates of metabolism by the recombinant CYPs to human liver microsomes. However, the normalized percentage data were related to inhibition data obtained with human liver microsomes (percentage of inhibition in the presence of CYP form-selective chemical inhibitors).
HPLRC.
Routine sample analysis was performed using HPLRC, with a Hewlett-Packard 1050 liquid chromatography system coupled to a Radiomatic FLO-ONE®\Beta model A-500 liquid scintillation flow detector (Machinist et al., 1995). Separations were achieved at ambient temperature with a SynChropak SCD-100 column (5 μm, 4.6 × 250 mm; Synchrom, Lafayette, IN). A SynChropak SCD-100 packed Javelin guard column was connected in series before the analytical column. Two mobile phases were used for the analyses. Mobile phase I consisted of 10% (v/v) acetonitrile/90% 0.05 M ammonium acetate (pH 4.6), whereas mobile phase II contained 70% (v/v) acetonitrile/30% 0.05 M ammonium acetate (pH 4.6). After injection of the sample (50–200 μl), a linear gradient was run from 60% (v/v) mobile phase I/40% mobile phase II to 100% (v/v) mobile phase II in a period of 15 min. The flow rate was maintained at 1 ml/min. Under these conditions, the retention times (±0.5 min) of ABT-761 and ABT-515 (hydroxy-ABT-761) were 12.5 and 9.0 min, respectively. ABT-438 (retention time, 13.5 min) and its corresponding hydroxy metabolite (retention time, 10 min) were similarly separated.
In the case of LC/MS analysis, the HPLRC unit was linked in tandem to a Perkin-Elmer Sciex API 300 triple-quadrupole mass spectrometer (Sciex, Toronto, Canada), which was equipped with a pneumatically assisted ion-spray source. Collisionally induced dissociation was performed using nitrogen as the collision gas. The HPLRC effluent was split 4:1, such that a 50-μl/min flow was directed into the mass spectrometer and the remainder was directed into the FLO-ONE®\Beta model A-500 liquid scintillation flow detector, which was equipped with a 75-μl flow cell. The ratio of column effluent to liquid scintillation fluid was 1:3.3. ABT-761 methylene bridge hydroxylation was confirmed, because the pseudo-molecular ion ([M+H]+) of ABT-761 (m/z 319) was distinct from that of ABT-515 (m/z 335; M + 16 amu). The pseudo-molecular ion of ABT-438 (m/z 302) was similarly distinguishable from that of its corresponding hydroxy metabolite (m/z 318; M + 16 amu).
Results
CYP-Dependent Metabolism of ABT-761 and ABT-438.
Typical radiochromatographs of the supernatants after incubation of [14C]ABT-761 and [14C]ABT-438 with human liver microsomes, in the presence of the NADPH-generating system, are presented in fig.2. After incubation with [14C]ABT-761, one major metabolite peak was observed; the metabolite coeluted with authentic ABT-515 and was identified by LC/MS as the methylene bridge hydroxy metabolite of ABT-761 (see Materials and Methods). A second peak (retention time, 15.5 min) was identified as an [14C]ABT-761 impurity (fig. 2A). MS analysis confirmed that ABT-438 also underwent methylene bridge hydroxylation, and a proposed scheme for the oxidative metabolism (stereochemistry unspecified) of ABT-761 and ABT-438 is shown in fig.1. There was no evidence that ABT-761 or ABT-438 underwent sulfoxidation and/or ring hydroxylation.
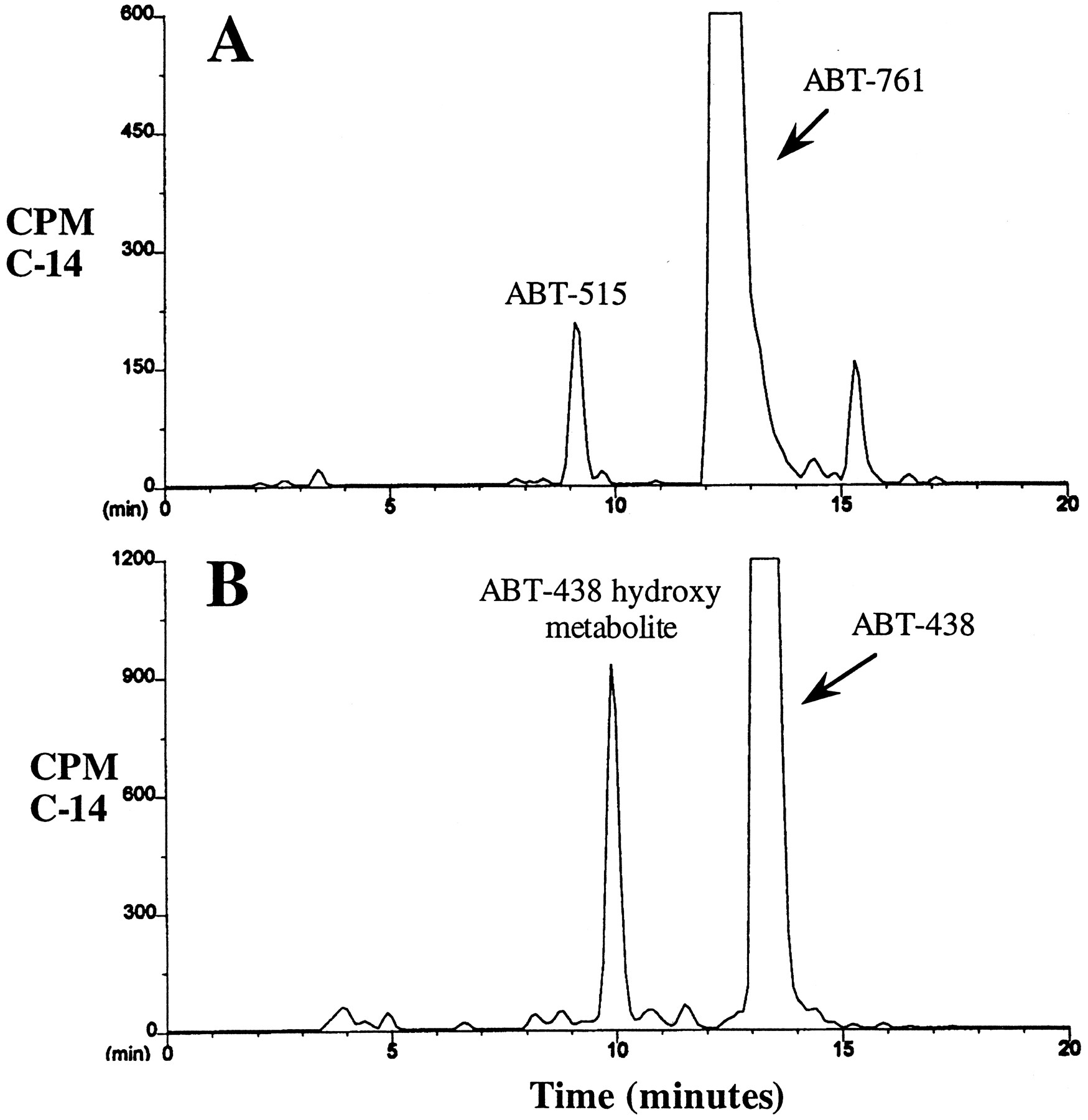
HPLC radiochromatograms for ABT-761 and ABT-438 after incubation with human liver microsomes.
[14C]ABT-761 (A) or [14C]ABT-438 (B) was incubated (at a final concentration of 10 μM) in the presence of human liver microsomes, as described in Materials and Methods.
Omission of the NADPH-generating system completely abolished the hydroxylation reaction with human liver microsomes, indicating that the process was enzymatic and NADPH-dependent. In addition, clotrimazole (50 μM), a potent nonspecific CYP inhibitor, markedly inhibited (>80%) the NADPH-dependent hydroxylation of both compounds, suggesting that the reaction was mediated by CYP (data not shown).
In a panel of human liver microsomes (N = 11), the interindividual variability in the rate of ring hydroxylation ranged from 4.8-fold with ABT-761 (range, 3.4–16.6 pmol/min/mg) to 7.0-fold with ABT-438 (range, 25.3–177 pmol/min/mg). Based on a comparison of mean specific activities in the panel of human microsomes, the rate of hydroxylation of ABT-438 was 9.2-fold greater than that of ABT-761 (78vs. 8.5 pmol/min/mg). Despite this variability, the rate of ABT-761 hydroxylation was highly correlated (r = 0.99,p < 0.001) with the rate of ABT-438 hydroxylation in the panel of human liver microsomes (table1).
Correlation of various CYP-selective monooxygenase activities with the hydroxylation of ABT-761 and ABT-438 in a panel of human liver microsomes
Reaction Kinetics.
To define the differences in the rates of hydroxylation of ABT-761 and ABT-438, an attempt was made to determine apparentKM and Vmaxvalues. However, over the concentration range studied (1–300 μM), the hydroxylation of both compounds did not conform to saturable Michaelis-Menten kinetics. Because of limitations in solubility, it was not possible to study reaction rates at higher concentrations (>0.3 mM). Because saturation was not achieved, the apparentKM could not be estimated from these data (fig. 3). Comparison ofVmax/KM ratios, however, indicated that the in vitro hepatic intrinsic clearance (mean ± SD, N = 3 different livers) for ABT-438 (1.7 ± 0.8 μl/min/mg) was approximately 10-fold greater than that for ABT-761 (0.17 ± 0.06 μl/min/mg).

Effects of substrate concentration on the rate of hydroxylation of ABT-761 and ABT-438.
The rates (v) of ABT-761 (A) and ABT-438 (B) hydroxylation were measured at different concentrations of drug ([S]). Because [S] ≪KM, intrinsic clearance (Vmax/KM) values were obtained from the slope of vvs. [S].
Correlation Studies.
ABT-761 and ABT-438 hydroxylation was significantly correlated with total CYP content (r = 0.92, p < 0.001, N = 11), erythromycin N-demethylase (CYP3A) activity (r = 0.83, p < 0.01,N = 11), COU hydroxylase (CYP2A6) activity (r = 0.73–0.74, p < 0.01,N = 11), and (S)-(+)-mephenytoin 4′-hydroxylase (CYP2C19) activity (r = 0.86–0.87,p < 0.001, N = 11) (table 1). In all instances, correlations were not influenced by a single outlying point. A statistically significant correlation (p < 0.001, N = 11) was also observed when the hydroxylation of ABT-761 and ABT-438 was correlated with the CYP3A/CYP2C9-dependent sulfoxidation of zileuton and ABT-193 (r = 0.97–0.98). In contrast, the correlation of zileuton/ABT-193 hydroxylation with ABT-761/ABT-438 hydroxylation was not statistically significant (r < 0.56, N = 11). The only other statistically significant (p < 0.05) correlations were obtained between ABT-761 hydroxylase and CYP2C9-selective tolbutamide hydroxylase (r = 0.63,N = 10) and CYP2D6-selective dextromethorphanO-demethylase (r = 0.60, N = 11) activities (table 1). By comparison, correlations with CYP1A2 and CYP2E1 activities were weak (r < 0.32,N = 11).
Inhibition Studies.
Ketoconazole (2.0 μM), a selective inhibitor of CYP3A at low concentrations, effectively decreased (by 55–70%) the rates of hydroxylation of ABT-761 and ABT-438 (fig.4). Similar results were obtained with troleandomycin (data not shown). Sulfaphenazole and tienilic acid, both CYP2C9 inhibitors, decreased hydroxylase activity by 10–20%. In contrast, marginal inhibition (≤10%) was observed with furafylline (CYP1A2-selective), quinidine (CYP2D6-selective), 4-methylpyrazole (CYP2E1-selective), and COU (CYP2A6-selective).

Inhibition of ABT-761 and ABT-438 metabolism, in the presence of human liver microsomes, by a series of CYP-selective inhibitors.
Data represent the mean ± SD of three different human livers. Inhibition data are expressed relative to activities measured in the presence of solvent alone. The final concentration of each inhibitor is indicated. ABT-761 (A) or ABT-438 (B) was incubated at a final concentration of 10 μM (≪KM). The proposed CYP form selectivity of the inhibitors was as follows: furafylline (FURA), CYP1A2; ketoconazole (KTZ), CYP3A; sulfaphenazole (SLF), CYP2C9; tienilic acid (TA), CYP2C9; quinidine (QND), CYP2D6; 4-methylpyrazole (4MP), CYP2E1; COU, CYP2A6.
Although α-naphthoflavone is a potent inhibitor of CYP1A2, it has been reported to stimulate several CYP3A-associated activities (Machinist et al., 1995; Shimada et al., 1989;Schwab et al., 1988). In the presence of α-naphthoflavone (10 μM), the rates of ABT-761 and ABT-438 hydroxylation were also increased 8-fold (8.0 ± 3.2 vs. 1.1 ± 0.2 pmol/min/mg, mean ± SD, N = 3 livers) and 4-fold (96 ± 39 vs. 25 ± 6.9 pmol/min/mg), respectively, providing evidence to implicate CYP3A forms as major components in the hydroxylation of both compounds (data not shown). In addition, (S)-(+) mephenytoin (0.5 mM, 10·KM for CYP2C19) failed to inhibit ABT-761/ABT-438 hydroxylase activity and actually stimulated activity (ABT-761, 3-fold; ABT-438, 2-fold) (data not shown).
Metabolism by cDNA-Expressed CYP Forms.
Of the CYP forms tested, CYP3A4 exhibited high rates of ABT-761 (1.0 pmol/hr/pmol of CYP) and ABT-438 (4.7 pmol/hr/pmol of CYP) hydroxylase activity (table 2). Relatively high rates of activity (0.6–1.3 pmol/hr/pmol) were also observed with microsomes containing cDNA-expressed CYP2C9, CYP2C19, or CYP2D6. By comparison, the rates of hydroxylation in the presence of cDNA-expressed CYP1A2, CYP2A6, or CYP2E1 were low (≤0.2 pmol/hr/pmol of CYP). No activity was detected in (control) microsomes prepared from B lymphoblastoid cells containing the selectable plasmid vector without a cDNA insert (table2).
Metabolism of ABT-761 and ABT-438 in the presence of human B lymphoblastoid microsomes containing cDNA-expressed CYP proteins
To obtain meaningful information, the turnover rates obtained with the various cDNA-expressed CYP proteins were normalized with respect to the nominal abundance of each CYP protein in native human liver microsomes (Shimada et al., 1994; Lecoeur et al., 1994), although it must be appreciated that the microsomal milieu differs in the two systems. It is apparent from the data presented in table 2 that the hydroxylation of ABT-761 and ABT-438 is mediated largely by CYP3A (66–86%), whereas a lesser contribution (9–20%) is made by CYP2C9. In accordance with the inhibition studies (fig. 4), the contributions of other CYP forms, such as CYP1A2, CYP2A6, CYP2C19, CYP2E1, and CYP2D6, are minimal (≤10%).
Discussion
The results of these studies demonstrate that ABT-761 and ABT-438 undergo CYP-dependent methylene bridge hydroxylation in the presence of human liver microsomes, and regression analysis indicates that hydroxylation of the two compounds is mediated by the same CYP form(s). Hydroxylation of ABT-761 by human liver microsomes is not unexpected, because ABT-515 has been detected in the urine of subjects receiving [14C]ABT-761 and in liver slice incubations with [14C]ABT-761 (Machinist JM, unpublished observations). For ABT-761 and ABT-438, the apparentKM values characterizing the hydroxylation by human liver microsomes exceeded the maximal concentration of drug tested (0.3 mM). This implies that the hydroxylation of ABT-761 and ABT-438 is characterized by a relatively highKM (≥0.3 mM). In agreement with this finding, ABT-761 and ABT-438 were shown to be relatively weak inhibitors (IC50 ≥ 0.2 mM,Ki ≥ 0.1 mM) of terfenadine hydroxylase (CYP3A) and tolbutamide hydroxylase (CYP2C9) activities (Rodrigues AD, unpublished observations).3
The results of this study also indicate that reduction of theN-hydroxy moiety of ABT-761, to form ABT-438, removes the site of direct glucuronidation and results in the formation of a metabolite that is a more efficient CYP substrate. Similar results were observed with zileuton and its N-dehydroxylated metabolite, ABT-193 (Machinist et al., 1995). Interestingly, thein vitro intrinsic clearance of ABT-761 (0.17 μl/min/mg) is similar to that of zileuton (0.08 μl/min/mg), despite the fact that the clearance of orally administered zileuton is greater than that of ABT-761 in vivo (493 vs. 29 ml/min) (Awni W, unpublished observations). Admittedly, binding of ABT-761 to human plasma proteins is greater than that of zileuton (99% vs.93%) (Machinist JM, unpublished observations). However, preliminary evidence suggests that the N-hydroxy glucuronidation of ABT-761 in vitro is characterized by a relatively low intrinsic clearance (compare these results with those for zileuton) (Bouska J, unpublished observations).
Several lines of evidence, obtained using correlation analyses, CYP form-selective inhibitors, and cDNA-expressed CYP proteins, have demonstrated that members of the CYP3A subfamily (most likely CYP3A4) are the principal human liver microsomal enzymes involved in the hydroxylation of ABT-761 and ABT-438 (≤10 μM). However, it was not possible to evaluate the role of CYP3A5 (vs. CYP3A4), because no attempt was made to measure the level of this enzyme in the microsomes used in this study. An ancillary role for CYP2C9 was demonstrated by the inhibition (13–20%) of hydroxylation in the presence of sulfaphenazole or tienilic acid, which was confirmed with the appropriate cDNA-expressed enzyme (table 2). However, a significant correlation with CYP2C9-selective tolbutamide hydroxylase activity (r = 0.63, p < 0.05, N= 10) was observed only in the case of ABT-761. The correlation between ABT-438 and tolbutamide hydroxylase activity, although relatively good (r = 0.61, N = 10), was not statistically significant.
Collectively, the data indicate that the hydroxylation of ABT-761 and ABT-438 is mediated by CYP3A and CYP2C9, although CYP2B6 was not included in the analysis. The CYP profiles are similar to those for zileuton and ABT-193, which both undergo CYP3A/CYP2C9-catalyzed sulfoxidation (Machinist et al., 1995). This was confirmed with the observation that the hydroxylation of ABT-761/ABT-438 was significantly correlated (r = 0.97–0.98,p < 0.001, N = 11) with the sulfoxidation of zileuton/ABT-193 (table 1).
In contrast to that of ABT-193 and zileuton, CYP1A2 does not play a major role in the metabolism of ABT-761 and ABT-438 (Machinist et al., 1995). For instance, the correlation between ABT-761/ABT-438 hydroxylation and CYP1A2-selective 7-ethoxyresorufinO-deethylase activity was not significant (r< 0.4, N = 11), and minimal inhibition (≤7%) was observed in the presence of furafylline, a CYP1A2-selective mechanism-based inhibitor (Machinist et al., 1995; Rodrigueset al., 1996). Moreover, the rate of hydroxylation of ABT-761 by cDNA-expressed CYP1A2 was least 10-fold lower than that observed with zileuton (≥0.8 vs. 0.1 pmol/hr/pmol), and the correlation between ABT-761/ABT-438 hydroxylation and CYP1A2/CYP2C9-mediated zileuton/ABT-193 hydroxylation was relatively weak (r = 0.52–0.56, N = 11) and not statistically significant. These differences may partly explain why ABT-761 does not elicit a clinically relevant drug-drug interaction with theophylline (Wong et al., 1998). By comparison, coadministration of zileuton to healthy subjects results in a doubling of theophylline AUC values (Granneman et al., 1995). This finding is clinically relevant, because the N-demethylation and C8-oxidation of theophylline are mediated largely by CYP1A2 and theophylline is characterized by a relatively narrow therapeutic index range (Campbell et al., 1987; Fuhret al., 1992; Sarkar et al., 1992; Gu et al., 1992). Similarly, zileuton has been shown to bring about a statistically significant increase (22%) in the AUC of (R)-warfarin, which is also a CYP1A2 substrate (Awniet al., 1995c; Rettie et al., 1992; Zhanget al., 1995).
Regression analysis indicated that ABT-761/ABT-438 hydroxylation and CYP2D6 (dextromethorphan O-demethylase) activity were significantly correlated (r = 0.6, p < 0.05, N = 11). However, correlation with CYP2D6 apoprotein levels was relatively weak (r = 0.50,N = 11), and quinidine failed to inhibit ABT-761 hydroxylase activity in native human liver microsomes (fig. 4). Similarly, the significant correlations with (S)-(+)-mephenytoin 4′-hydroxylase (CYP2C19) and COU 7-hydroxylase (CYP2A6) activities are considered fortuitous, because no appreciable inhibition of ABT-761/ABT-438 hydroxylase activity (≤1%) was observed in the presence of (S)-(+) mephenytoin or COU. Correlation with these activities is inevitable, because (S)-(+)-mephenytoin 4′-hydroxylase activity correlates with erythromycin N-demethylase activity (r = 0.81, p < 0.01, N = 11) and COU 7-hydroxylase activity correlates with the level of total CYP (r = 0.88, p < 0.001,N = 11) in our panel of microsomes (Rodrigues AD, unpublished observations).
In conclusion, ABT-761 and its N-dehydroxylated metabolite (ABT-438) are metabolized via CYP3A- and CYP2C9-dependent methylene bridge hydroxylation in the presence of native human liver microsomes. Other CYP forms (e.g. CYP2D6, CYP2E1, CYP1A2, CYP2C19, and CYP2A6) do not contribute significantly to the metabolism of either compound. Therefore, ABT-761 differs from zileuton, which undergoes CYP1A2/CYP2C9-catalyzed ring hydroxylation and CYP3A/CYP2C9-mediated sulfoxidation (Machinist et al., 1995). The clinical relevance of these findings will ultimately depend on the fraction of the ABT-761 dose that is metabolized viaoxidative metabolism, because both CYP2C9 and CYP3A have been shown to be induced by various agents (e.g. phenytoin, rifampicin, rifabutin, phenobarbital, and/or carbamazepine) (Guengerich, 1995). Because ABT-761 (maximal concentration of drug in plasma, ≤18 μM), like zileuton (Machinist et al., 1995), is a relatively weak inhibitor of CYP-dependent monooxygenase activityin vitro (IC50 ≥ 0.2 mM), it is expected that the CYP3A/CYP2C9 drug-drug interaction profile for ABT-761 would be similar to that for zileuton. For instance, zileuton has been shown to elicit marginal effects (≤20% increase in AUC) on the pharmacokinetics of drugs such as (S)-warfarin, phenytoin, prednisone, and naproxen (Awni et al., 1995a,b,c;Samara et al., 1995). Moreover, some of these drugs (e.g. naproxen) are also known to undergo direct glucuronidation (Vree et al., 1993).
Acknowledgments
We thank Dr. R. Lee (Abbott Laboratories) for performing LC/MS analyses. We are also indebted to S. Thomas (Abbott Laboratories) for purifying [14C]ABT-761 and [14C]ABT-438.
Footnotes
-
Send reprint requests to: J. M. Machinist, Ph.D., Department 46V, Building AP9, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, IL 60064.
-
↵1 Present address: Drug Metabolism, Merck Research Laboratories, P.O. Box 4, WP26A 2044, Sumneytown Pike, West Point, PA 19486-0004.
-
↵3 Inhibition studies were carried out at a defined substrate concentration ([S] ≈ KM). Assuming competitive inhibition, for [S] ≈KM, IC50/2 ≈Ki. In addition,KM ≈ Kifor a competitive inhibitor (cosubstrate).
- Abbreviations used are::
- CYP
- cytochrome P450
- COU
- coumarin
- HPLRC
- high performance liquid radiochromatography
- Received January 8, 1998.
- Accepted May 12, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}