


Abstract
Efavirenz (Sustiva) is a potent and specific inhibitor of the HIV-1 reverse transcriptase and is approved for the treatment of HIV infection. The metabolism of efavirenz in different species has been described previously. Efavirenz is primarily metabolized in rats to the glucuronide conjugate of 8-OH efavirenz. Electrospray ionization-liquid chromatography/mass spectrometry analyses of bile samples from rats dosed with either efavirenz or with 8-OH efavirenz revealed three polar metabolites, M9, M12, and M13, with pseudomolecular ions [M-H]− atm/z 733, 602, and 749, respectively. The characteristic mass spectral fragmentation patterns obtained for metabolites M9 and M13 suggested that these were glutathione-sulfate diconjugates, and the presence of a glutathione moiety in metabolite M9 was confirmed by liquid chromatograpy/nuclear magnetic resonance (NMR) analysis of bile extracts. Metabolite M12 was characterized by liquid chromatography/mass spectrometry as a glucuronide-sulfate diconjugate. Unambiguous structures of M9, M12, and M13 were obtained from one-dimensional proton and carbon NMR as well as proton-proton (correlated spectroscopy, two-dimensional shift correlation), proton-carbon heteronuclear multiple quantum correlation, and long-range proton-carbon (heteronuclear multiple bond correlation) correlated two-dimensional NMR analyses of metabolites isolated from rat bile. The mass spectral and NMR analyses of M10, which was isolated from rat urine, suggested a cysteinylglycine-sulfate diconjugate. The isolation of these polar metabolites for further characterization by NMR was aided by mass spectral analyses of HPLC fractions and solid phase extraction extracts during the isolation steps. The complete characterization of these novel diconjugates demonstrates that further phase II metabolism of polar conjugates such as sulfates could take place in vivo.
The effective treatment of HIV infection and AIDS is still difficult despite tremendous advances in our understanding of the pathogenesis of the disease and the arrival of potent drugs aimed at different, critical targets in the life cycle of the virus (Havlir and Richman, 1996). Clearly, optimal treatment involves multiple drug therapy designed to decrease the viral burden as low as possible. New agents with convenient dosing regimens are needed to ensure compliance. Efavirenz (Fig. 1) is a potent non-nucleoside inhibitor of the HIV-1 reverse transcriptase. Clinical trials have demonstrated a durable, long-lasting reduction in HIV RNA after once-a-day dosing in combination with other drugs (Staszewski et al., 1998). To more fully understand the disposition of this agent in relevant species for safety assessment, the metabolism of efavirenz has been described (Mutlib et al., 1998a,b).

Chemical structure of efavirenz.
Rats, an important species for safety assessment studies, metabolize efavirenz extensively. The glucuronide conjugate of 8-OH efavirenz was found as the major metabolite in urine of rats dosed with efavirenz (Christ et al., 1997; Mutlib et al., 1998a,b). In addition to this glucuronide conjugate, a number of polar diconjugates were found in the urine and bile of rats dosed with either efavirenz or with 8-OH efavirenz. These polar metabolites were isolated from urine and bile by solid-phase extraction (SPE) and chromatography on reversed-phase HPLC columns. Often the structures of very polar metabolites are not elucidated due to the difficulty in isolating and separating them from endogenous components in the biological matrices. Characterization of these polar conjugates can give very useful information regarding the metabolic pathways and evidence for any potentially reactive metabolites of a compound. This article describes the identification of novel, mixed diconjugates resulting from oxidation and conjugation with glucuronic acid, sulfate and glutathione using liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS), liquid chromatography/nuclear magnetic resonance (LC/NMR), and NMR approaches. These metabolites are biologically novel and formation of these types of metabolites may represent a previously unappreciated combination of drug metabolism reactions.
Materials and Methods
Chemicals and Supplies.
(S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one (efavirenz,C14H9ClF3NO2;mw of 315.68); (S)-6-chloro-4-(cyclopropylethynyl)-8-hydroxy-4-(trifluoromethyl)-1,4-dihydro-2H-3,1-benzoxazin-2-one(M4or8-OHefavirenz, C14H9ClF3NO3,mw of 331.68); and (S)-6-chloro-4-(cyclopropylethynyl)-8-sulfo-4-(trifluoromethyl)-1,4-dihydro-2H-3,1-benzoxazin-2-one (M3 or 8-OH efavirenz sulfate, C14H9ClF3NO6S,mw of 411.74) were synthesized and characterized by the DuPont Pharmaceuticals Company (Markwalder et al., 1998). Amberlite XAD-2 and glutathione were purchased from Sigma Chemical Co. (St. Louis, MO). Bond-Elut C18 cartridges (10 g/60 ml) were obtained from Varian Sample Preparation Products (Harbor City, CA). All general solvents and reagents were the highest grade available commercially.
LC/MS.
LC/MS was carried out by coupling a Waters HPLC system to a Sciex API 300 mass spectrometer. The HPLC eluent was introduced into the source using a turbo ionspray interface held at 400–480°C. The electrospray needle was maintained at 4000 to 5000 V with the orifice potential set at 50 to 60 V. The nebulizer gas was ultrapure nitrogen set at 40 p.s.i. The turbo ionspray gas flow rate was 6 liters/min. The mass spectrometer was operated in the negative ion mode (Q1 and MS/MS) to detect the diconjugates. MS/MS was carried out using nitrogen as the collision gas. The collision energy was kept at 30 to 40 eV.
For the analyses of bile samples, HPLC was carried out using a Waters quarternary pump (model 616) coupled in sequence to a Waters WISP (model 717 plus) and to a Beckman C18 column (250 × 4.6 mm, 5 μm). The metabolites were separated by a gradient solvent system consisting of a mixture of acetonitrile and 10 mM ammonium formate, pH 3.5. The percentage of acetonitrile was increased from 25 to 80% in 20 min. For the analyses of rat urine, HPLC was done on the same column (250 × 4.6 mm, Beckman) using a gradient solvent system consisting of a mixture of acetonitrile and 0.1% acetic acid. The percentage of acetonitrile was increased from 25 to 80% in 20 min using a solvent flow rate of 1.0 ml/min. Aliquots of bile and urine samples were injected directly onto the columns. A postcolumn split (1:1) introduced approximately 0.5 ml/min of eluent to the mass spectrometer.
To detect the metabolites, aliquots of the fractions from XAD-2 column, C18 cartridges and from semipreparative HPLC column were introduced to the mass spectrometer using flow injection analyses method. The mobile phase consisted of 30% methanol in 0.1% acetic acid delivered at a rate of 0.25 ml/min.
LC/NMR.
Liquid chromatography/proton nuclear magnetic resonance (1H-LC/NMR) was performed using a Bruker AMX-500 MHz NMR spectrometer equipped with a dedicated1H flow-probe (probe flow cell of 4 mm i.d. with a volume of 120 μl). Stopped-flow 1H-NMR spectra were obtained at 500 MHz using a modified one-dimensional version of the nuclear Overhauser effect spectroscopy pulse sequence for solvent peak presaturation, which produced conditions for ensuring double solvent suppression. Stopped-flow spectra were acquired using 256 or 512 transients with 64K data points and a spectral width of 12,000 Hz. HPLC was performed using a Bruker LC22C pump and LC313 variable wavelength detector. The outlet of the UV detector was connected to the HPLC-NMR probehead via an inert polyethylether ketone capillary (0.25 mm i.d.). HPLC was performed on a C18 column (250 × 4.6 mm, 5 μm) using a gradient elution consisting of two components: (A) 0.1% TFA (deuterated) in D2O and (B) acetonitrile (Pestnal/analytical grade, Riedel deHaen, Germany). The gradient consisted of increasing the percentage of Bfrom 18 to 30 in 10 min followed by another increase to 40%B in 5 min. The flow rate was set at 1.0 ml/min.
High-Field NMR.
The purified metabolites, M9, M10, M12, and M13 were dissolved in methanol-d4 and filtered to remove particulate matter. The structures of these metabolites (approximately 1–2 mg of each) were determined from proton and carbon 1-dimensional NMR as well as proton-proton [correlated spectroscopy, two dimensional shift correlation (COSY)], proton-carbon heteronuclear multiple quantum correlation (HMQC), and long-range proton-carbon [heteronuclear multiple bond correlation (HMBC)] correlated two-dimensional NMR experiments using a 400 MHz Varian VXR-400 instrument.
Animal Studies.
Ten male Sprague-Dawley rats with cannulated bile ducts (weighing between 250–400 g) were dosed orally with efavirenz (50 mg/ml, 0.5% methylcellulose solution) once daily at 250 mg/kg for 3 days, then twice daily for the following 7 days. Serial bile and urine samples were collected over ice and pooled from all the animals on a daily basis and stored frozen at −20°C until analyzed. The dosing volume was 5 ml/kg. To elucidate the secondary metabolic pathways, another study was done in which six female and three male bile duct-cannulated rats were given 8-OH efavirenz (250 mg/kg, p.o.) and the sulfate conjugate of 8-OH efavirenz (80 mg/kg, i.v.), respectively. The urine and bile samples were collected over ice for 24 h.
Isolation of the Diconjugates (Metabolites M9, M12, and M13) from Rat Bile.
Bile obtained from several rats was pooled, diluted 1:1 with distilled water, and loaded onto an XAD-2 column (300 × 25 mm). The sample was allowed to elute under gravity at a rate of less than 1 ml/min. After the sample had been loaded, the column bed was washed with 2 × 150 ml of deionized water (pH ∼5–6) followed by elution with 100% methanol (200 ml). Aliquots from both the aqueous and the organic fractions were analyzed by LC/MS. The fractions containing the polar metabolites (M9, M12, and M13) were subsequently loaded onto C18 cartridges (10 g/60 ml) for further purification. The cartridges were eluted with increasing percentages of methanol in water (5–100% methanol) and the eluent was assayed for metabolites by LC/MS. The fractions containing the three metabolites, M9, M12, and M13, were pooled and dried under vacuum. The dried residues were reconstituted in water and rechromatographed on the C18 cartridges using different proportions of methanol in water as the eluent. After drying the samples, the extracts were purified on a semipreparative HPLC column as described below.
Semipreparative HPLC separation of the polar metabolites isolated from bile was carried out on a Waters Symmetry C18 column (7.8 × 300 mm, 7 μm) using methanol/2% acetic acid (65:35 v/v) as the eluent. The flow rate was 3.5 ml/min. The eluent was monitored at 254 nm using a variable wavelength detector (Waters 486). Three peaks were collected corresponding to metabolites M9 (m/z 733), M12 (m/z 602), and M13 (m/z749). Samples corresponding to each metabolite were pooled and dried under vacuum. Final purification was done by rechromatographing each isolated metabolite on C18 cartridges. After pooling the fractions containing the metabolites (each fraction from C18 analyzed by LC/MS before pooling), the samples were dried under vacuum and submitted for NMR analyses. Approximately 1 to 2 mg of each metabolite, M9, M12, and M13, were isolated for NMR analyses.
Isolation of the Cysteinylglycine Conjugate (Metabolite M10) from Rat Urine.
Urine samples from rats dosed with efavirenz were pooled and extracted on C18 cartridges (10 g/60 ml) using between 5 to 50% of methanol in water as the eluent. The fractions containing metabolite M10 were pooled and dried under vacuum. The dried residues were reconstituted in 2% acetic acid and rechromatographed on C18 cartridges. The metabolite was eluted with 55 to 60% methanol from the cartridge. After drying, the extracts were further purified by semipreparative HPLC on a Beckman C18 column (10 × 250 mm, 5 μm) using an isocratic HPLC system. The dried extract obtained from the C18 cartridge was reconstituted in methanol/water (1:4, v/v) and rechromatographed on the column using acetonitrile/0.1% acetic acid (27:73, v/v) at a flow rate of 5 ml/min as the eluent. The peak corresponding to metabolite M10 was collected after multiple injections, dried, and repurified on the same column using methanol/acetonitrile/0.1% acetic acid (9:28:63, v/v) as the mobile phase. The metabolite was collected, dried under vacuum, and rechromatographed on a C18 cartridge. The fractions containing the metabolite were pooled, dried, and submitted for NMR analyses. Approximately 4 to 5 mg of M10 was isolated using this procedure.
Synthesis and Isolation of Efavirenz Glutathione Adducts.
Efavirenz (100 mg) and reduced glutathione (120 mg) were dissolved in 50 ml of tetrahydrofuran and stirred at room temperature for 3 days after the addition of 50 μl of triethylamine. At the end of the reaction, tetrahydrofuran was dried under vacuum and the residue reconstituted in 50 ml of water. The mixture was extracted with 2 × 50 ml of ethyl acetate to remove the unreacted efavirenz and the aqueous phase chromatographed on a C18 cartridge. The desired glutathione adducts were eluted from the cartridge with 70% methanol in water. The sample was dried and chromatographed on a semipreparative C18 HPLC column (Beckman, 10 × 250 mm). The glutathione adducts were separated using a gradient HPLC consisting of solvent A (100% acetonitrile) and solvent B (0.1% acetic acid). The percentage of solventA in the mobile phase was maintained at 25% for the first 15 min and increased to 30% in the following 10 min. The solvent was delivered at 4 ml/min. This gradient ensured optimal resolution between the two glutathione adducts (minor and major peaks). The peak corresponding to the major glutathione conjugate was collected, pooled, dried under vacuum, and submitted for mass spectral and NMR analyses.
Results
LC/MS and NMR Characterization of Polar Diconjugates in Rat Bile.
Metabolites M9, M12, and M13 were found in the bile of male and female rats dosed with either efavirenz, 8-OH efavirenz (M4) or with the sulfate conjugate of 8-OH efavirenz (M3). The three biliary metabolites, M9, M12, and M13 with pseudomolecular ions ([M-H]−) at m/z 733, 602, and 749, respectively (Figs. 2-4), were isolated in sufficient quantities for unambiguous confirmation of the structures by NMR experiments.
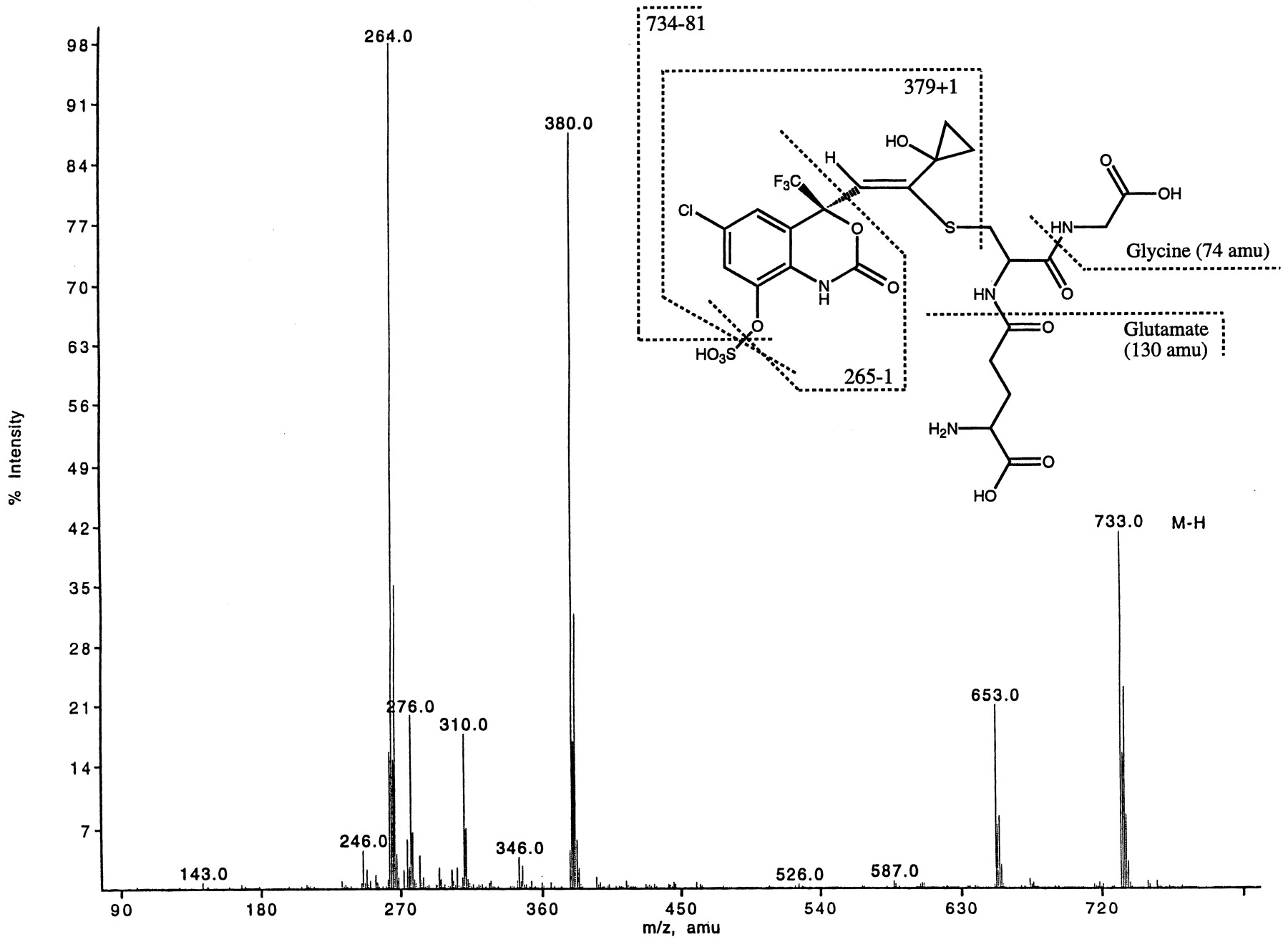
Metabolite M9 was found to be a glutathione-sulfate diconjugate of dihydroxylated efavirenz. M9 showed a characteristic loss of a sulfate group (−80 amu) from the parent ion ([M-H]−at m/z 733). The total addition of 419 amu to the molecule suggested that this metabolite was produced as a result of initial hydroxylation(s) and subsequent conjugations carried out by phase I and II enzymes, respectively. Because a sulfate group was present, the mass difference (419–80 = 339) suggested the addition of two oxygen atoms (phase I oxidation) as well as a glutathione moiety (32 + 307 = 339). Figure 2 shows an ion atm/z 380 indicative of a fragment ion produced by cleavage of the sulfur-carbon bond of the glutathione adduct. The MS/MS spectrum of metabolite M9 showed that the glutathione moiety was attached to the cyclopropyl ethynyl side chain.1H-LC/NMR of metabolite M9 present in rat bile provided first conclusive evidence of a glutathione adduct and the possible site of conjugation. The 1H-LC/NMR showed two signals for the aromatic ring [δ 7.52 and 7.08, which were coupled to each other by total correlated spectroscopy (TOCSY)], a singlet at δ 6.40 (an alkene proton), and multiplets at δ 0.95 and 1.08, which were assigned to the cyclopropane protons. The characteristic multiplet for the cyclopropyl methine proton seen in1H-NMR of efavirenz (at approx. δ 1.5) was absent (as was demonstrated by TOCSY experiments). The glutathione protons were assigned by TOCSY experiment: δ 4.15 (cys α), 3.73 (gly α), 3.68 (glut α), 3.41 (cys β), 3.02 (cys β′), and 2.32 (glut β). The other two –CH2– protons of glutamate were buried under a solvent peak. The one-dimensional1H-NMR of the isolated metabolite clearly showed the presence of the glutathione, the loss of the aromatic proton on C8, and the appearance of a new singlet at 6.4 ppm (Fig.3). The COSY showed the expected correlations among the protons. Using COSY, the lack of correlations with the C15 and C16 protons, also confirmed that the methine proton on C14 was absent. The HMQC and HMBC provided information on the position of conjugation with the glutathione as well as allowing a full assignment of the protonated carbons. Of particular importance was the carbon (δ at 122 ppm) bearing the proton at 6.4 ppm. The large chemical shift of this carbon at 122 ppm indicated that this was a vinylic carbon. The signal at 122 ppm was assigned to C12. This assignment was also supported by the correlations of the C12 proton at 6.4 ppm to C11, C9, C4, and C14. The HMBC results showed that cys-β protons had a strong correlation to a quarternary carbon at 153 ppm. Hence the glutathione moiety was attached to the molecule via the carbon-sulfur link to carbon at 153 ppm. The protons on C15 and/or C16 showed a correlation into the carbon at 58.2 ppm, thus, this carbon was assigned as C14. The chemical shift of C14 suggested that this carbon also bore an electronegative group. From the mass spectral data, this group was either a hydroxyl or a hydrogen sulfate. Comparisons with reference materials (1H-NMR of the synthetic sulfate conjugate of 8-OH efavirenz) suggested that the hydrogen sulfate was at the C8 position and not on C14. The large chemical shift of the carbon at 153 ppm indicated that this was a vinylic carbon having an additional deshielding from the thio-ether. The common correlation of the cys-β and the C15/C16 protons to carbon at 153 ppm made it possible to assign this as C13. The geometry of the double bond was confirmed by the nuclear Overhauser effect difference NMR. The structure of metabolite M9 is shown in Fig. 2 and Scheme FS1.

Electrospray ionization (ESI)-LC/MS of metabolite M9 ([M-H]− at m/z 733) isolated from rat bile.

1H-NMR of metabolite M9 (m/z 733) isolated from bile of rats dosed with efavirenz.

Proposed metabolic pathways for the formation of mixed diconjugates of efavirenz in rats.
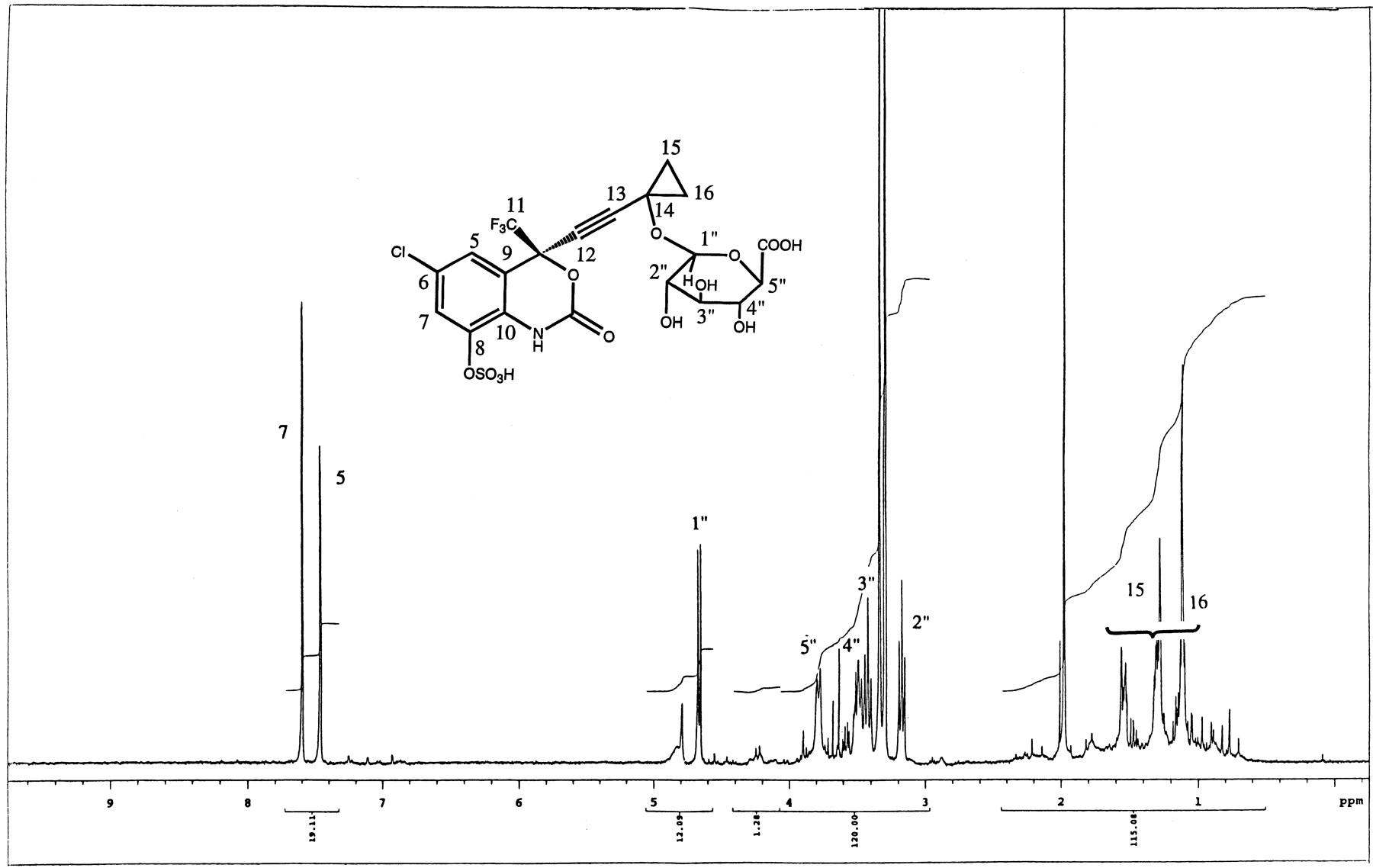
Metabolite M12 was identified as a glucuronide-sulfate diconjugate of dihydroxylated efavirenz. The mass spectral data of M12 showed characteristic losses of 80 and 176 amu (Fig.4) from the parent ion ([M-H]− at m/z 602), indicating a diconjugate of dihydroxylated efavirenz (314 + 32 + 80 + 176). MS/MS of the parent ion (m/z 602) produced characteristic fragment ions at m/z 522 and 426, confirming losses of sulfate and glucuronic acid moieties, respectively. The fragment ion at m/z 264 indicated that the second hydroxyl group (and the glucuronic acid) was probably on the cyclopropyl ethynyl side chain.1H-NMR data (Fig.5) showed the existence of two aromatic protons consistent with hydroxylation at C8. The methine proton at C14 was distinctly absent as shown by COSY experiments. Two-dimensional NMR was used to determine the sites of conjugation with glucuronic and sulfuric acids on the molecule. The glucuronic acid was attached at C14 as demonstrated by HMBC experiments that showed that the anomeric proton of the glucuronic acid was correlated to C14. The assignment for C14 was made by correlation between this carbon and C15 and C16 protons. The C15 and C16 protons also showed correlation with the quaternary carbon at 92 ppm that was assigned to the C13 carbon.

ESI-LC/MS of metabolite M12 ([M-H]− at m/z 602) isolated from rat bile.
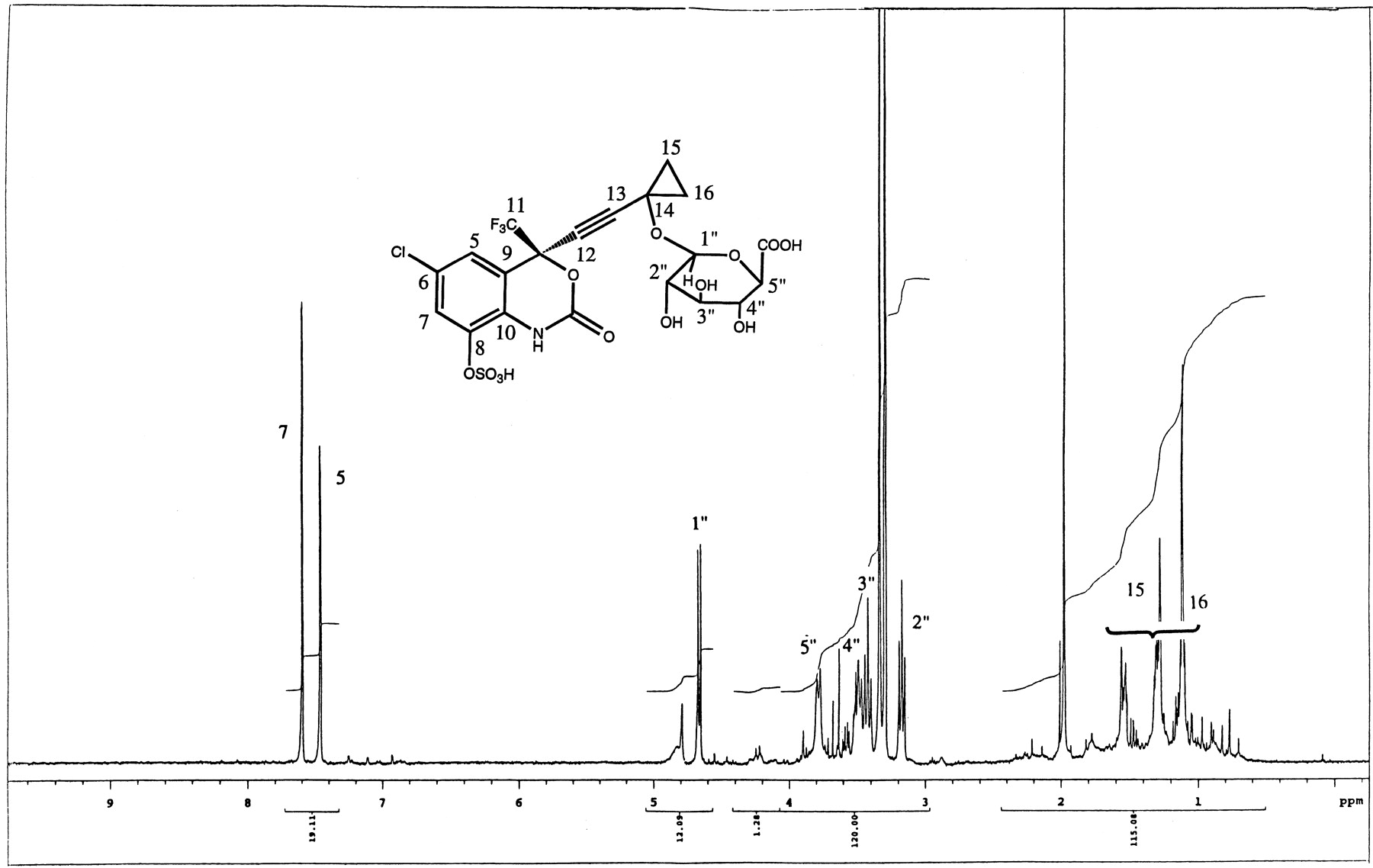
. 1H-NMR of metabolite M12 (m/z 602) isolated from bile of rats dosed with efavirenz.
Metabolite M13 was identified as an α,β-unsaturated ketone with glutathione and sulfate groups attached at C13 and C8, respectively. M13 also showed characteristic loss of 80 amu (Fig.6) from the parent ion ([M-H]- at m/z 749). The pseudomolecular ion indicated an addition of 16 amu to metabolite M9. The MS/MS spectrum of metabolite M13, however, showed a different fragmentation pattern compared with that of metabolite M9. The presence of glutathione was indicated by the fragment ion atm/z 306. The ion at m/z 396 was produced as a result of retention of sulfur on the aglycone after the cleavage of carbon-sulfur bond (see Fig. 6). Consecutive losses of two molecules of water from m/z 396 gave ions atm/z 378 and 360. The hydroxylated aromatic ring system (similar to M9) was characterized by a fragment ion atm/z 264. The MS/MS data, however, did not provide sufficient information to elucidate the complete structure of metabolite M13. Metabolite M13 was also isolated from rat bile and its structure confirmed by NMR experiments. The1H-NMR data (Fig.7) clearly showed that the cyclopropyl protons were absent, indicating that the cyclopropyl group was no longer intact. The chemical shift of the C16 protons indicated a hetero-atom attached at that position. The mass spectral data suggested a hydroxyl group at C16. The vinylic proton on C12 of metabolite M13 was shifted further downfield as compared with metabolite M9. This is probably due to the deshielding caused by the carbonyl group at C14. The glutathione protons are assigned as shown in Fig. 7. The carbon shifts of C12, C13, and C14 are consistent with an α-β unsaturated ketone. Correlations from C15 and C16 protons to C14 confirmed this assignment.
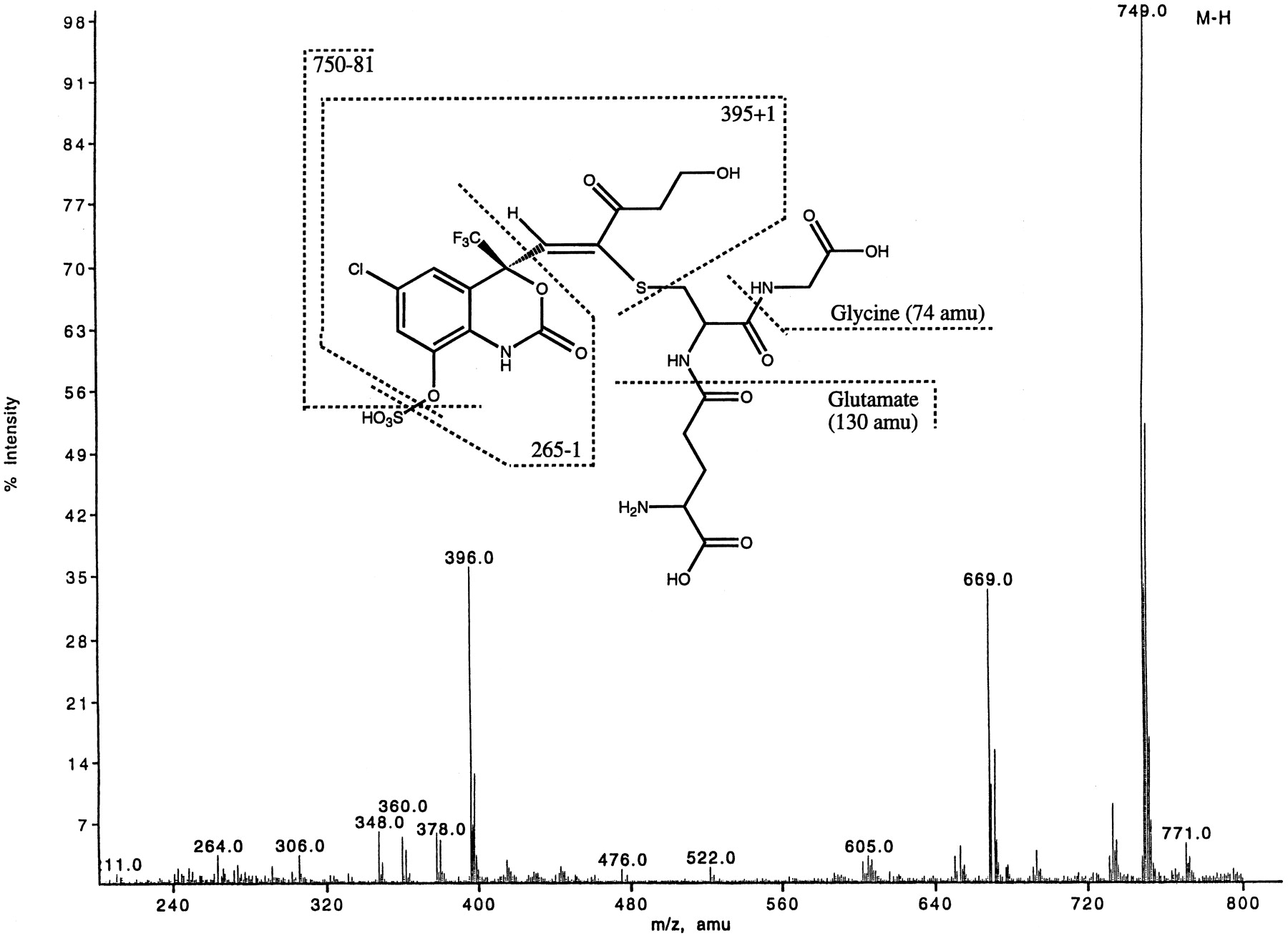
ESI-LC/MS of metabolite M13 ([M-H]− at m/z 749) isolated from rat bile.

1H-NMR of metabolite M13 (m/z 749) isolated from bile of rats dosed with efavirenz.
Diconjugates in Rat Urine.
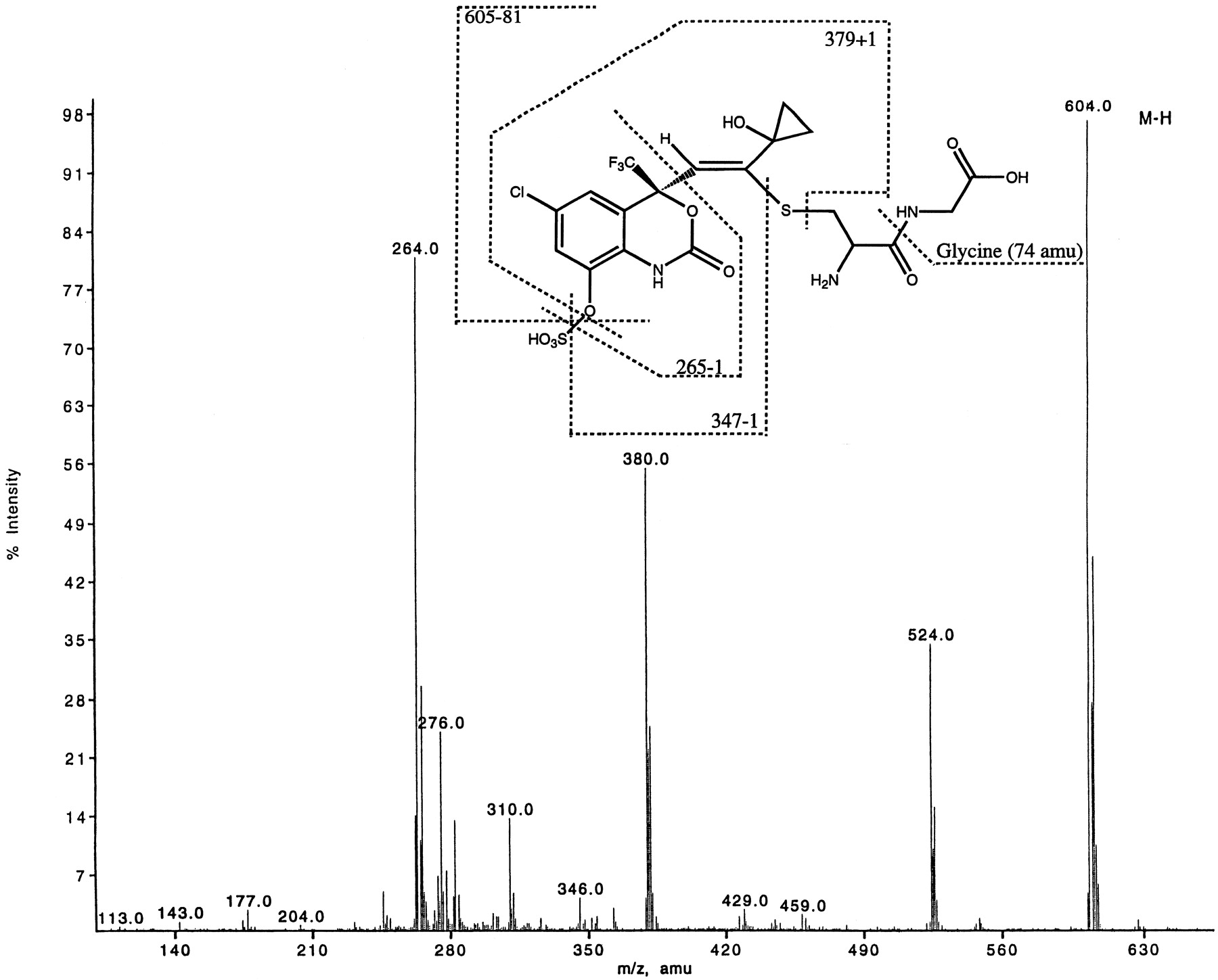
Metabolite M10, isolated from rat urine, was identified as a cysteinylglycine-sulfate diconjugate with [M-H]− at m/z 604. It showed a similar fragmentation pattern (Fig.8) as metabolite M9; however M10 had a mw 129 amu less than that of metabolite M9. Fragment ions atm/z 380 and 264 confirmed that this was aS-linked metabolite probably formed by the hydrolysis of glutamate (−129 amu) from metabolite M9. The structure of metabolite M10 was confirmed by isolating sufficient quantities from rat urine and carrying out one-dimensional proton and carbon NMR, gradient-enhanced correlated spectroscopy, gradient-enhanced HSQC, and gradient-enhanced HMBC two-dimensional NMR analyses. The 1H-NMR of M10 is shown in Fig.9. The chemical shifts of C12, C13, C14, C15, and C16 were similar to the corresponding carbons in metabolite M9. The major difference between the 1H-NMR spectra of the two metabolites (M9 and M10) was the absence of any resonances from the glutamic acid for M10 (compare Figs. 3 and 9). The long-range correlations from the cys-β protons to C13 confirmed the point of attachment of the cysteinyl-glycine moiety. This was also confirmed by the long range correlations from the C12 protons to the carbons at C4 and C9.

ESI-LC/MS of metabolite M10 ([M-H]− at m/z 604) isolated from rat urine.

1H-NMR of metabolite M10 (m/z 604) isolated from urine of rats dosed with efavirenz.
In addition to M10, metabolite M12 was also found in rat urine. M9 was detected in trace quantities in urine of rats given higher doses of efavirenz (Mutlib et al., 1998b).
Synthetic Glutathione Conjugate.
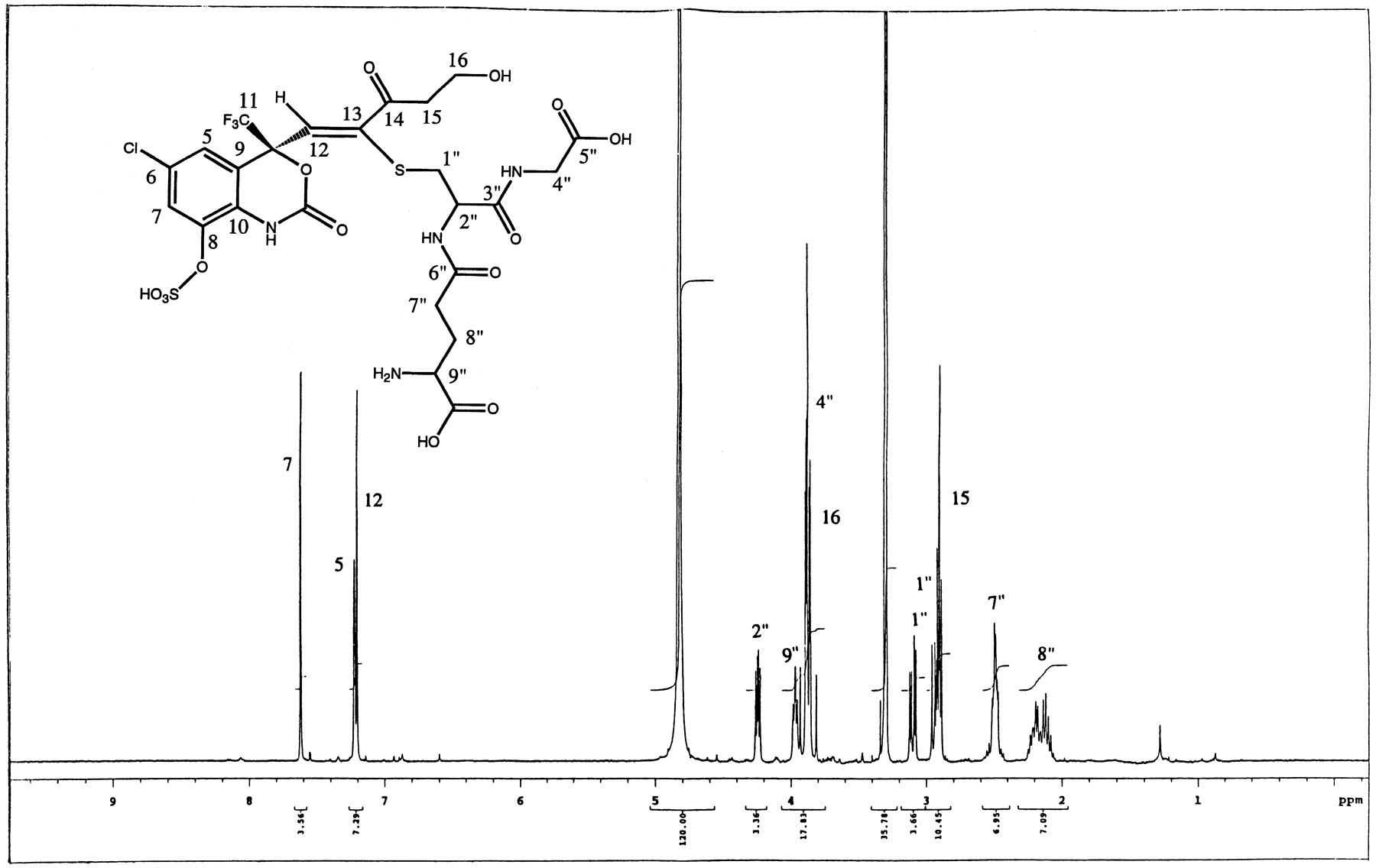
Even though the direct conjugation of efavirenz with glutathione was never demonstrated from in vivo studies, an attempt was made to chemically synthesize standards of possible glutathione conjugates of efavirenz. The synthetic glutathione adducts of the parent separated as a minor and a major peak on HPLC system gave identical mass spectral data with [M-H]− at m/z621. The MS/MS fragment ions of the major component gave ions atm/z 128, 143, 179, and 210 indicative of the glutathione moiety. The NMR spectra showed addition of the glutathione moiety on C12 versus C13 (as was seen with metabolites M9, M10, and M13). The 1H-NMR data also demonstrated that the cyclopropyl ring was cleaved during the addition process (Fig.10). An allene structure was proposed based on NMR results. The carbon shift of C14 and the coupling pattern between the three aromatic ring protons was found to be consistent with the proposed structure. The structure of the synthetic glutathione conjugate was confirmed by one-dimensional proton, gradient-enhanced correlated spectroscopy and gradient-enhanced HSQC two-dimensional NMR.
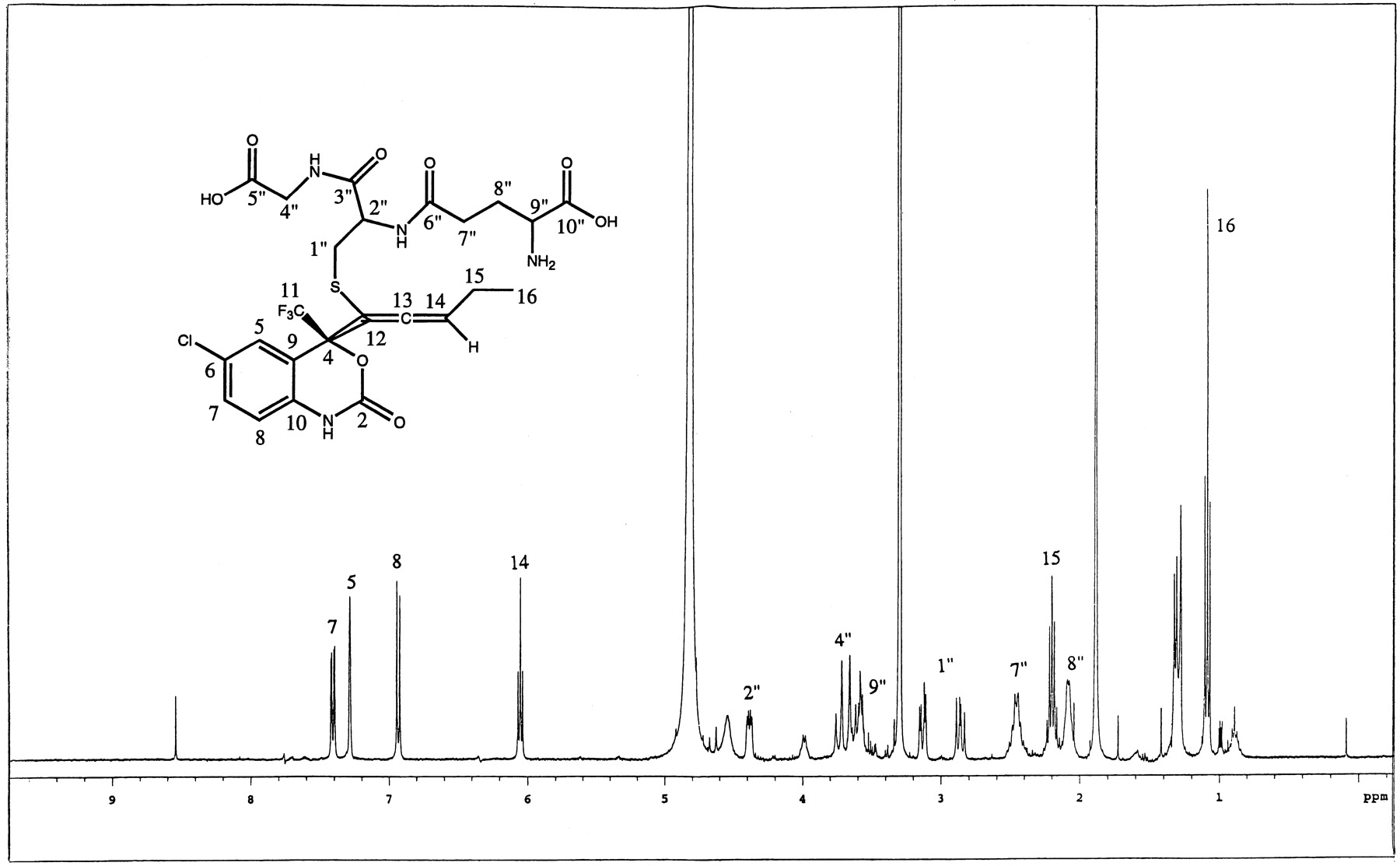
1H-NMR of synthetic glutathione adduct of efavirenz.
Discussion
Numerous reports have appeared in the literature describing the mass spectrometric and NMR characterization of monoconjugates such as glucuronides, sulfates, and glutathione adducts of compounds (Lehmann et al., 1982; Liberato et al., 1983; Weidolf et al., 1988; Draper et al., 1989; Muck and Henion, 1990; Mutlib and Abbott, 1992; Burlingame et al., 1992). There are, however, only a limited number of reports on spectroscopic characterization of diconjugates. The diglucuronides of endogenous bile components (Blumenthal et al., 1977; Blanckaert, 1980) and disulfates of steroids such as estriol and estrone (Levitz et al., 1974; Hobkirk et al., 1977) have been previously described. Full spectroscopic data (including MS and NMR spectroscopy) have not been presented for any of these diconjugates. Recently a report appeared in the literature describing the mass spectrometric and1H-NMR characterization of a glutathione-glucuronide polar diconjugate of valproic acid metabolite (Tang and Abbott, 1996). These diconjugates are inherently difficult to isolate from endogenous components, hence formation of diconjugates remain a poorly documented metabolic route for xenobiotics.
The characterization of diconjugates of efavirenz was facilitated by the presence of chlorine M + 2 isotope clusters in the mass spectra. This made it easy to trace drug-related compounds in complex biological matrices such as bile. By using LC/MS, three polar diconjugates (metabolites M9, M12, and M13) of efavirenz were isolated from rat bile and characterized further by MS/MS and NMR. The isolation of these very polar metabolites for further characterization by NMR was aided by mass spectrometric analyses of HPLC fractions and SPE extracts during the isolation steps. This ensured that appropriate samples containing the metabolites were collected for further purification on semipreparative HPLC, hence saving a tremendous amount of time. In the past, UV detection was the only means of detecting the presence of metabolites; however the nonspecific nature of this method made it difficult to isolate the desired metabolites. With the advent of LC/MS it has become very routine to focus on particular HPLC peaks or SPE fractions that contain the metabolites of interest. The use of LC/NMR in the recent years (Spraul et al., 1992; Mutlib et al., 1995; Shockcor et al., 1996;Lindon et al., 1997) has made it easier to characterize metabolites present in biological matrices. Nonetheless, to obtain full NMR characterization of some complex metabolites, isolation of these compounds from endogenous components may still be required. In this study LC/NMR was used to obtain initial information on the nature of metabolite M9. The 1H-LC/NMR analysis was not adequate for complete structural characterization of metabolite M9. The full characterization of this diconjugate was achieved only after purification of this metabolite from bile components. Of the three metabolites, metabolite M9 (m/z 733) was the most abundant component followed by metabolites M12 and then M13. These metabolites are formed after initial hydroxylations (at C8 and at C14), probably mediated by hepatic cytochrome P-450 enzymes, followed by subsequent conjugations with sulfuric acid and with either glucuronic acid or glutathione. All three metabolites were sulfated at the 8-OH position on the aromatic ring.
Metabolite M9 was a glutathione conjugate (see Scheme FS1 for the structure) found in the bile of rats dosed with either efavirenz, 8-OH efavirenz (M4) or the sulfate conjugate of 8-OH efavirenz (M3). The addition of glutathione took place across the triple bond of the cyclopropyl ethynyl side chain giving a trans configuration as demonstrated by the nuclear Overhauser effect difference NMR experiment. The attachment of glutathione via the sulfur linkage was at C13 adjacent to the carbon (C14) bearing the hydroxyl group. For comparison, a glutathione adduct of efavirenz was synthesized and its structure determined by mass spectral and NMR analyses. An adduct resulting from direct conjugation of efavirenz with glutathione in vivo has not been found in any of the species studied (Mutlib et al., 1999). The structural analysis of the synthetic glutathione adduct showed that the point of attachment was at C12 instead of C13. The NMR data indicated that on addition of glutathione to the triple bond, the cyclopropyl ring was opened (see Fig. 10) producing an allene conjugate. The nonenzymatic addition of GSH to C12 was very slow and the overall yield was less than 5%. The enzyme-catalyzed addition of GSH (see Mutlib et al., 1998a,b) on C13 as in metabolite M9 could be partly due to the activating effect of the hydroxyl group (another electron withdrawing substituent) on C14. The addition of a glutathione moiety to a triple bond activated by a hydroxyl group on an adjacent cyclopropyl ring is unique and has not been previously described.
Metabolite M12 was found both in bile and urine of rats dosed with either efavirenz or with 8-OH efavirenz. Metabolite M12, ([M-H]− at m/z 602), was identified as a glucuronide-sulfate diconjugate of dihydroxylated efavirenz with a mw of 347. The mass spectral data clearly confirmed the existence of both glucuronic and sulfuric acids on the molecule. The site of conjugation with glucuronic acid at C14 was confirmed by NMR experiments. The position of sulfation was the C8 phenol group (the other hydroxyl group that could be conjugated).
Metabolite M13, with a mw of 750 amu ([M-H]−at m/z 749), was identified as an α,β-unsaturated ketone with glutathione and sulfate groups attached at C13 and C8, respectively. The MS/MS fragmentation for this metabolite was different from that of metabolite M9. The NMR experiments carried out on isolated metabolite provided conclusive evidence for the structure of this compound (Scheme FS1). The NMR data showed that the cyclopropane ring was cleaved producing an α,β-unsaturated ketone. The α,β-unsaturated ketone is a Michael acceptor and could potentially interact with nucleophilic macromolecules in rat liver or bile.
Metabolite M10, which was found in urine of rats, is a product of γ-glutamyl transpeptidase cleavage of metabolite M9 (Mutlib et al., 1998b). Metabolite M10 was found to produce similar MS/MS spectrum as metabolite M9. The NMR data confirmed the absence of the glutamate moiety as expected. Metabolite M10 is unique because the cysteinylglycine conjugates have not, to our knowledge, been isolated from urine before. These products are believed to be intermediates formed during the normal catabolism of glutathione adducts (Commandeur et al., 1995 and the literature cited therein). These cysteinylglycine adducts are usually found in the bile of animals. The final breakdown products of glutathione conjugates are usually cysteine orN-acetylcysteine conjugates, which are normally excreted in urine. Metabolite M10 was found in the urine of rats in significant quantities after 250-mg/kg doses of efavirenz or 8-OH efavirenz. Further studies are being done to isolate sufficient quantities of this metabolite to quantitate the levels of this cysteinylglycine conjugate in the urine of rats. Analysis of urinary cysteinylglycine conjugate may provide valuable information about the extent and mechanism of bioactivation of efavirenz via glutathione conjugation in vivo.
The proposed metabolic pathways leading to the formation of the metabolites M9, M10, M12, and M13 are shown in Scheme FS1. An initial hydroxylation of efavirenz leads to the formation of 8-hydroxy efavirenz (metabolite M4) which in turn is further sulfated and hydroxylated to produce the cyclopropanol intermediate, M11. Analyses of urine and bile samples from rats dosed with metabolite M4 showed the presence of the same S-linked conjugates as were found in rats dosed with efavirenz. Results from the analyses of samples from rats dosed with the sulfate conjugate of metabolite M4 (now called metabolite M3) showed significant quantities of glutathione and cysteinylglycine conjugates in bile and urine, respectively. Hence, it is postulated that 8-OH efavirenz is formed first before further hydroxylation takes place on C14. Furthermore it appears that sulfation of M4 takes place before hydroxylation on the cyclopropyl ring because after dosing authentic 8-OH sulfate (M3), M9 was found as a significant metabolite in rat bile. The cyclopropanol metabolite, M11 (Mutlib et al., 1998a,b), then undergoes further biotransformation by conjugation with either glucuronic acid or with glutathione. Hydroxylations of compounds already possessing sulfate moieties have been previously described for a number of steroids including estrone 3-sulfate (Tsoutsoulis and Hobkirk, 1980), estrone-3-sulfate (Hobkirk et al., 1977), and estradiol 17β-sulfate (Watanabe et al., 1982). Watanabe and coworkers (Watanabe et al., 1991) demonstrated that cytochrome P-450 is responsible for catalyzing the hydroxylations of the aromatic ring of estradiol 17β-sulfate. Sulfate conjugates are not the only phase II conjugates capable of further secondary metabolism; glucuronides are also substrates of cytochrome P-450 or other phase II enzymes. Evidence for hydroxylation of estradiol-17β,17-glucuronide to produce 2-hydroxyestradiol-17β,17-glucuronide by rat liver microsomes has been presented (Watanabe and Yoshizawa, 1983). It was demonstrated that 2-hydroxylation took place without the cleavage of the conjugate group, indicating that estradiol-17β,17-glucuronide could act as a substrate for rat liver microsomal 2-hydroxylase. Similarly it was shown that estriol-3-glucosiduronate was a metabolic intermediate in the conversion of estriol to diconjugates by rabbits (Miyazaki et al., 1980). Tang and Abbott (1996) showed that a glucuronide conjugate of (E)-2-propyl-2,4-pentadienoic acid, a metabolite of valproic acid, was further conjugated with a glutathione to produce a glucuronide-glutathione diconjugate. In studies carried out in this laboratory, significant quantities of M9, M10, M11, M12, and M13 were all produced by rats administered the sulfate conjugate of M4 (data not shown). Because only trace quantities of 8-OH glucuronide conjugate was detected in bile and urine of these rats, it was postulated that M3 (the sulfate conjugate of M4) was directly hydroxylated to produce M11 rather than being deconjugated first followed by further hydroxylation. This has also been confirmed recently by in vitro metabolism studies in which M11 was produced from M3 in the presence of rat liver microsomes and NADPH (Mutlib et al., 1998b).
Enzymatic addition of glutathione across the triple bond of the cyclopropanol intermediate (M11) leads to the formation of metabolite M9. Recent in vitro studies demonstrated that a cytosolic glutathioneS-transferase was responsible for this reaction (Mutlib et al., 1998b). In rats, the cyclopropanol intermediate is also trapped as the glucuronide-sulfate diconjugate and excreted as metabolite M12 in urine and bile.
Metabolite M9 could be a precursor to metabolite M13. A further oxidation of the hydroxyl group at C14 could lead to the formation of a keto group with concurrent opening of the cyclopropane ring. Metabolism of a glutathione conjugate by further oxidation of the aglycone has not been previously described in the literature. Metabolite M9 could also be transported to kidney where it could be processed by enzymes γ-glutamyltranspeptidase, dipeptidases, and β-lyase, which are normally responsible for the degradation of glutathione adducts (Commandeur et al., 1995 and references cited therein). Significantly, metabolite M9 was hydrolyzed to metabolite M10 without any evidence of further degradation to mercapturic acids at doses up to 250 mg/kg of efavirenz. Metabolites M9 and M10 could also be produced by rat kidneys because this tissue contains significant levels of glutathioneS-transferase. The formation of metabolite M10 from M9 and the involvement of cyclopropanol metabolite (M11) as a substrate for glutathione transferases in rats is described elsewhere (Mutlib et al., 1998b).
The formation of these diconjugates involves both the cytochrome P-450 (phase I) and cytosolic/microsomal phase II enzymes (sulfotransferases, glucuronosyltransferases, and glutathioneS-transferases). The formation of metabolite M12 is interesting because the secondary sulfate metabolite (cyclopropanol intermediate M11, see Scheme FS1) must be transported back into the endoplasmic reticulum (where the glucuronosyltransferases reside) to produce the sulfate-glucuronide diconjugate. The formation of metabolite M9 from this cyclopropanol intermediate, as stated before, was found to be mediated by glutathione S-transferases present in the cytosol. The free cyclopropanol intermediate was not detected in rat urine.
These studies have demonstrated conclusively that polar diconjugates can be isolated with relative ease with the aid of MS for further characterization by NMR. Determining the structures of these metabolites gives insight into the metabolic pathways of formation and indirect evidence for the existence of potentially reactive intermediates that would not otherwise be detected. The results from these studies have also demonstrated that highly polar metabolites, such as sulfates, are also capable of being further metabolized by phase II enzymes leading to diconjugates.
Acknowledgments
LC/NMR experiments were conducted by M. Spraul and M. Hofmann at Bruker Analytische Messtechnik (Rheinstten, Germany). Efavirenz, 8-OH efavirenz, and its sulfate conjugate were supplied by S. Seitz and J. Markwalder of the DuPont Pharmaceuticals Company.
Footnotes
-
Send reprint requests to: Dr. A. E. Mutlib, Drug Metabolism and Pharmacokinetics Section, DuPont Pharmaceuticals Company, P.O. Box 30, 1094 Elkton Rd., Newark, DE 19714. E-mail address: abdul.mutlib{at}dupontpharma.com
- Abbreviations used are::
- LC/MS/MS
- liquid chromatography/mass spectrometry/mass spectrometry
- ESI-LC/MS
- electrospray ionization-liquid chromatography mass spectrometry
- SPE
- solid-phase extraction
- 1H-NMR
- proton nuclear magnetic resonance
- COSY
- correlated spectroscopy, two dimensional shift correlations
- TOCSY
- total correlated spectroscopy
- HMBC
- heteronuclear multiple bond correlation
- HMQC
- heteronuclear multiple quantum correlation
- Received March 15, 1999.
- Accepted June 15, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}