


Abstract
An existing cryopreservation method for liver slices applies 12% dimethylsulfoxide and rapid freezing. We found that cells in rat liver slices cryopreserved in this manner deteriorated rapidly upon culturing. To improve this cryopreservation method, we varied the dimethylsulfoxide concentration (0, 12, 18, and 30%), the cryopreservation medium (Williams medium E, fetal calf serum, and University of Wisconsin medium), slice thickness, and the storage period at 4°C during slice preparation before cryopreservation. After thawing, slices were cultured for 4 h at 37°C before their viability was evaluated by their potassium content and the number of intact cells determined histomorphologically. The biotransformation capacity of liver slices cryopreserved by the improved method was assessed by testosterone oxidation, hydroxycoumarin sulfation, and glucuronidation. Best results were obtained with 18% dimethylsulfoxide in Williams medium E: the potassium content of cryopreserved slices was higher than 65%, and the number of intact cells was higher than 60% of that in fresh slices; with 12% dimethylsulfoxide, potassium content was less than 40%, and the number of intact cells was less than 30%. Results did not differ between the three cryopreservation media. Viability of thin slices (8–10 cell layers) was better maintained than that of thicker slices (>14 cell layers). Storage at 4°C of slices before cryopreservation decreased viability after cryopreservation. Both oxidative and conjugation activities were better than 60% of fresh values. Although results varied, slices cryopreserved with this improved method and cultured for 4 h retained viability between 50 and 80%, and biotransformation activity between 60 and 90% of fresh slices.
Precision-cut tissue slices are extensively used as in vitro tool for studies on toxicity and biotransformation of xenobiotics (Bach et al., 1996; Olinga et al., 1997b). A major advantage of tissue slices over primary cell cultures is that cell-to-cell contacts and normal tissue micro-architecture remain intact. In addition, tissue slices are prepared according to a rapid, reproducible, and relatively simple method (Krumdieck et al., 1980). Cryopreservation of liver slices can extend their use, for slices from animals and man could then be used in a single experiment, thus facilitating species comparison and decreasing number of laboratory animals required.
A number of cryopreservation methods for liver slices has been described in recent years. Roughly, methods can be divided in those that use (computer-controlled) slow freezing (Fisher et al., 1991,1993, 1996), those that use vitrification protocols requiring high concentrations of cryoprotectants (Wishnies et al., 1991; Ekins, 1996;Ekins et al., 1996), and those that apply rapid freezing with low concentrations of dimethylsulfoxide (DMSO)1(de Kanter and Koster, 1995; Glöckner et al., 1996, 1998;de Kanter et al., 1998). Success of reported methods is variable and seems dependent on animal species (Fisher et al., 1991) and viability parameters (Maas et al., 2000). Particularly, phase I biotransformation is well preserved by all methods, while phase II biotransformation capacity was reported to be preserved to a much lesser extent or not at all (Wishnies et al., 1991; Ekins, 1996; Ekins et al., 1996; de Kanter et al., 1998; Maas et al., 2000). No publications have reported preservation of intact histomorphology of thawed liver slices.
The fast-freezing method for cryopreservation, developed in our laboratory (de Kanter and Koster, 1995; de Kanter et al., 1998), has the advantage of using relatively low and nontoxic concentrations of cryoprotectant (12% DMSO). Moreover, the method is relatively simple: slices are put in cryovials with medium and directly submersed in liquid nitrogen. Using this method, slices apparently maintained functional activity (testosterone oxidation, urea synthesis) and cell membrane integrity (alanine aminotransferase retention) close to levels of fresh slices in the early post-thaw stage (0–3 h after thawing). Others, who use fast-freezing methods have found similar results (Glöckner et al., 1996).
Recently, we compared the usefulness of this method, which presumably minimizes intracellular ice-crystal formation (IIF) by intracellular vitrification, to another method that was designed to prevent IIF by dehydrating cells by slow freezing (Maas et al., 2000). Although the fast-freezing method better maintained phase I biotransformation activity and urea synthesis, cell integrity as revealed by histomorphological examination was severely affected by either method. Moreover, cryopreserved slices had low contents of intracellular potassium (K+) and ATP and were incapable of performing GSH conjugation of chlorodinitrobenzene due to cofactor (GSH) loss.
In the present study, we attempted to improve results on slice viability after cryopreservation by fast-freezing. Two approaches were used for this purpose. One approach was to try to inhibit ice-crystal formation and thus stimulate vitrification of intracellular and extracellular water. To achieve this, we varied the concentration of DMSO, a well known glass former (Boutron and Kaufmann, 1978; Hey and MacFarlane, 1996; Baudot et al., 1999). Additionally, Williams medium E (WME) was replaced as cryoprotectant carrier medium by University of Wisconsin medium (UW) and fetal calf serum (FCS), media that contain hydroxyethyl starch and bovine serum albumin, respectively, that bind water and thus stimulate water vitrification extracellularly (Körber et al., 1982; Takahashi and Hirsh, 1985).
The other approach was to alter the susceptibility of slices for cryopreservation damage. To that end, slice thickness was varied. Slice thickness may be of importance, because ice-crystal formation is more likely to occur in the cryopreservation medium surrounding the slices during freezing than in the tissue itself, where vitrification is favored (Takahashi and Hirsh, 1985; Peridieu et al., 1995). In very thin slices, therefore, relatively more cells might suffer from ice-crystal formation on the outside of the cell than in thicker slices. In very thick slices, on the other hand, cryoprotectant diffusion to the inner cell layers may be impeded causing a suboptimal cryopreservation condition in these cell layers.
In tissues kept at subphysiological temperatures, intracellular ion concentrations change and cells swell (Belzer and Southard, 1988). Inevitably, slices are kept for some time in buffer at 4°C before they enter the cryopreservation procedure. Therefore, the duration of this step was tested for its effect on the cryopreservation result.
As endpoints for optimization of viability, K+retention and slice histomorphology were used, parameters that have been shown to be highly sensitive to cryopreservation damage (Maas et al., 2000). Slices, cryopreserved by an improved protocol, were tested on their phase I (testosterone oxidation) and phase II biotransformation capacity (hydroxycoumarin sulfation and glucuronidation conjugation) subsequently.
Experimental Procedures
Materials.
7-Hydroxycoumarin glucuronide and sulfate were generous gifts of Dr. P. Olinga, Center for Pharmacy, University of Groningen, The Netherlands; Krebs Henseleit buffer, insulin, gentamycin were obtained from Sigma, Axel, The Netherlands; WME, FCS, and phosphate-buffered saline were from Gibco BRL, Breda, The Netherlands; DMSO (>99.9% pure), cryovials were from Greiner, Alphen a/day Rijn, The Netherlands; Coomassie Protein kit 23200, was from Pierce, Oud-Beyerland, The Netherlands; testosterone and 11β-hydroxytestosterone were from Fluka, Buchs, Switzerland; hydroxytestosterone metabolites were from Steraloids Inc., Wilton, NH; 7-hydroxycoumarin was from Merck-Schuchart, Hohenbrunn, Germany; hematoxylin and eosin (H&E) were from Sigma, St. Louis, MO.d-Glucose, HPLC-water, and all other chemicals were from Baker, Deventer, The Netherlands.
Preparation of Slices.
Male rats (Wistar, obtained from Harlan CPB, Horst, The Netherlands) had free access to food and water. They were anesthetized with 35% CO2, 65% O2 before the livers were removed. Tissue cores (8 mm) were prepared from the freshly excised liver, using an electrical drill (Metabo BSE 5010) with a tissue coring tool (Alabama R&D, Munford, AL). Slices were prepared subsequently using a Krumdieck tissue slicer (Alabama R&D), filled with oxygenated (95% O2, 5% CO2) ice-cold Krebs-Henseleit buffer, supplemented with NaHCO3 (25 mM) and CaCl2 (2.5 mM). To determine optimal slice thickness for cryopreservation, the slicer was set to produce slices of various thicknesses. The thickness of slices generated in this manner was evaluated by microscopically counting the number of cell layers in paraffin cross-sections.
After preparation, slices were washed and kept on ice in WME with 10% FCS until use. To determine the influence of this cold storage period on slice viability after cryopreservation, the length of time between slice preparation and the start of the cryopreservation method was varied in some experiments.
Cryopreservation and Thawing of Slices.
Before freezing, slices were preincubated in a shaking water bath (110 times/min) for 30 min in a 25-ml Erlenmeyer flask (6 slices/Erlenmeyer) on ice. Each Erlenmeyer flask contained 5 ml of oxygenated medium (WME, UW, or FCS) supplemented with cryoprotectant. As cryoprotectant, various concentrations of DMSO were used. Subsequently, slices were put in 2-ml cryovials with 0.5 ml of preincubation medium. Hereafter, the cryovials were directly submersed in liquid nitrogen. Following this procedure, slices were cooled at approximately 250°C/min to −196°C (de Kanter and Koster, 1995). After storing the cryopreserved slices in liquid nitrogen for 30 min2 or more, slices were thawed by placing the cryovials in a 37°C water bath. After visible ice was vanished, slices were washed briefly with WME, 10% FCS.
To determine possible adverse effects of preincubation with the cryoprotectants on the slices, part of the slices were preincubated with the cryoprotectant but not cryopreserved. The cryopreservation medium was washed away immediately at the end of preincubation, whereafter slices were incubated for viability testing.
Incubation of Slices.
Fresh and cryopreserved slices were incubated for 4 h at 37°C, floating in 5 ml of medium in a 25-ml Erlenmeyer flask (1 slice/Erlenmeyer), placed in a water bath shaking back and forth (110 times/min), under humid carbogen (95% O2, 5%CO2). This incubation system has proven to maintain viability of fresh liver slices for at least 24 h (Olinga et al., 1997a). WME was used as incubation medium, supplemented with FCS (5%), 0.1 μM insulin, 50 mg/liter gentamycin andd-glucose (to a medium concentration of 25 mM). After the 4 h incubation period, tests on viability and biotransformation capacity were performed.
K+ Content.
To determine K+ slice content, slices were washed in a physiological concentration of NaCl (37°C) in K+-free HPLC water. Subsequently, the slice was cut in two equal parts. One part was used for histomorphological examination (see below). The other part was immersed in 70% ethanol in HPLC water, containing 2 mM EDTA (pH 10.9), for K+ and protein determination. For this purpose, the slice was homogenized using a Branford sonifier (50% duty cycle, 5 s). K+ content was measured in the homogenate using a Beckman 2 electrolyte analyzer (Beckman Instruments-Netherlands Division BV, Mijdrecht, The Netherlands).
Protein Determination.
To an aliquot of the slice homogenate, 2 M NaOH (25% v/v) was added to dissolve the protein. Hereafter, the protein solution was diluted (1:5 minimally) with phosphate-buffered saline. Subsequently, protein content was measured by a Cobas-Bio centrifugal spectrophotometer with a Coomassie Protein kit, bovine serum albumin was used as a standard.
Histomorphological Examination.
After fixation with 70% ethanol at 4°C for at least 24 h, slices were dehydrated in a series of ethanol and xylene using a Shandon 2LE Processor. Subsequently, slices were vertically embedded in paraffin and cross-sections (4-μm thick) were made, which were stained with H&E. On microscopical examination, slice viability was determined by estimating the percentage of viable cells in the slice cross-section. For determining viability, nuclear shape and staining and cytoplasmatic staining was taken into account.
Testosterone Metabolism.
Metabolism of testosterone was determined after 60 min of incubation with 250 μM testosterone in homogenate extracts by HPLC as earlier described but using a flow of 0.8 ml/min (van‘t Klooster et al., 1993). 11β-Hydroxytestosterone was used as internal standard. Of all testosterone metabolites peak areas were determined, but only formation of 2α-hydroxytestosterone (an important hydroxytestosterone metabolite in rats) was quantified (as production in picomoles per slice per minute). The formation of this metabolite was linear throughout the 60-min incubation period with the substrate, with R2 = 0.999.
7-HC Conjugation.
Conjugation of 7-HC to 7-HC-glucuronide (7-HC-gluc) and 7-HC-sulfate (7-HC-sulf) was determined after incubation of 60 min with 100 μM 7-HC. The conjugates were determined in the slice homogenate by HPLC on and Inertsil ODS-3 (150 × 2.1 mm) with a gradient from 100% water with 3 g/liter ammonium acetate to 100% pure methanol in 30 min and 3 min isocratic at 100% methanol. Detection was at 325 nm (Walsh et al., 1995). Formation of 7-HC- glucuronide and 7-HC-sulfate was linear throughout the 60-min incubation period with the substrate with R2 = 0.995 and 0.998, respectively.
Statistical Analysis.
For the variables cryoprotectant concentration, cryopreservation medium and storage time, an ANOVA has been applied to the viability data, considering data of one experiment as one observation, followed by comparisons between data with the t test. A Bonferroni correction of the P values was used where applicable. To study the effect of slice thickness, an ANOVA has been applied for the fresh and cryopreserved slices separately. Statistical significance of the relation (both with a linear and a quadratic fit) between viability and slice thickness was subsequently determined with the F test. For all tests, P < .05 was considered statistically significant.
Results
Histomorphology of precision cut liver slices.
Precision cut liver slices always have damaged cells at the cutting edges (1–2 cell layers at each edge). These damaged cells are also taken into account with viability scoring. That means that 20 to 30% of the cells are always nonviable and, therefore, slices will get a maximal “viability” score of 70 to 80%.
Effect of DMSO on the Viability of Fresh Slices.
To discriminate the possible toxicity of DMSO used in the cryopreservation procedure from damage caused by cryopreservation itself, the effect of DMSO on the viability of fresh slices was determined. To that end, freshly prepared slices were subjected to the cryopreservation preincubation with DMSO and, without actual cryopreservation, cultured for 4 h at 37°C.
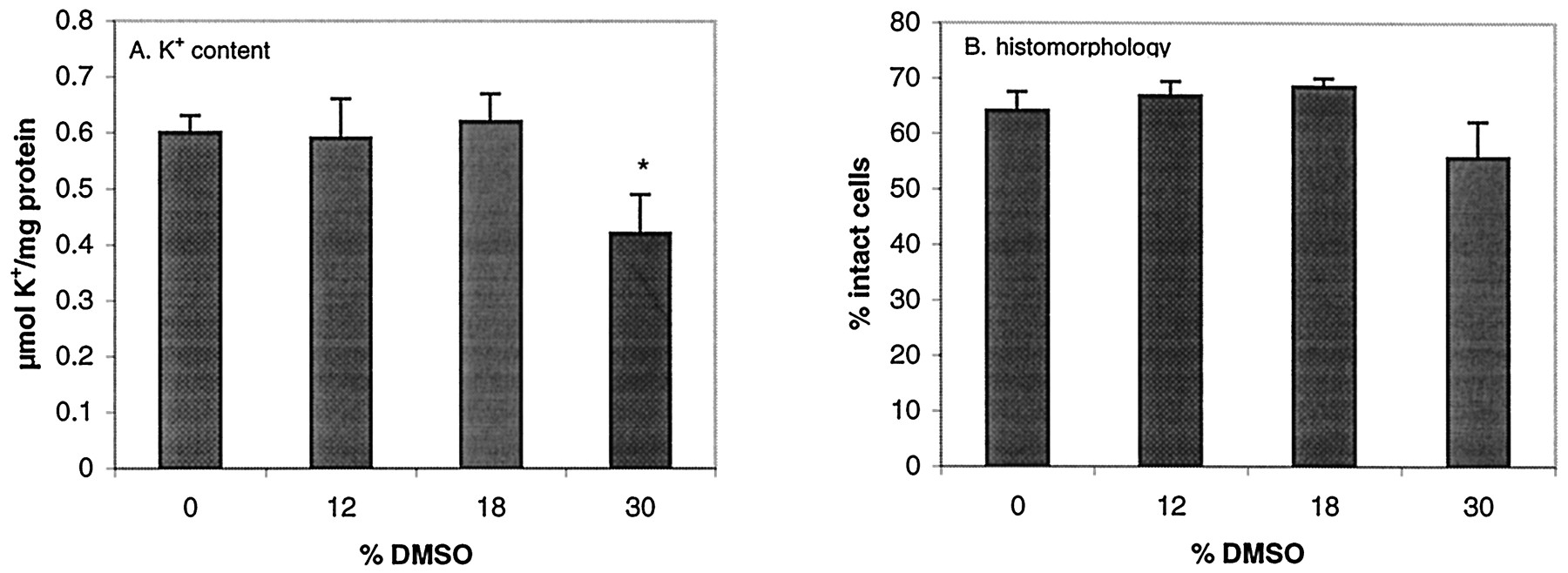
Exposure of slices to DMSO at concentrations up to 18% during preincubation (not followed by cryopreservation) did not cause adverse effects on slice K+ content or on slice histomorphology (Fig. 1). The use of 30% DMSO resulted in a statistically significant reduction in K+ content of fresh liver slices and a nonstatistically significant reduction in intact cells.

K+ content (A) and histomorphology (% intact cells) (B) in fresh slices in culture for 4 h after preincubation with various concentrations of DMSO.
Values are shown as mean of individual experiments (six to nine experiments, 3 slices per experiment) + S.E.M. ∗, values are significantly lower (P < .05) than those in slices that were preincubated without DMSO.
Cryoprotection by DMSO.
Immediately after thawing, K+ concentration and the number of intact cells in cryopreserved slices were comparable to fresh slices, independent of the DMSO concentration used (12–30%) (data not shown). After 4 h of culturing, however, 18% (v/v) DMSO better retained viability of slices than 12 and 30% (Fig.2). The K+ content of slices cryopreserved with 18% DMSO was over 65% of the content of fresh slices (i.e., slices that were not preincubated with DMSO or cryopreserved), and higher than the results with 12 or 30% DMSO (40 and 55%, respectively). Histomorphological examination gave similar results. The number of intact cells in slices cryopreserved with 18% DMSO was over 60% of that of fresh slices and with 12 and 30%, this was considerably lower (Fig. 2A). Without DMSO, only a few cells were intact, and in most cells pyknosis and karyorhexis was observed (Fig. 3A), whereas with 12% DMSO, a number of cells with intact nuclei can still be seen. Slices cryopreserved with 18%, the aspect of the cells (Fig. 3D) is hardly distinguishable from that in a fresh slice (Fig. 3A).

K+ content (A) and histomorphology (% intact cells) (B) in cryopreserved slices preincubated with various concentrations of DMSO.
Slices were cultured for 4 h after thawing. Values are shown as mean of individual experiments (four to ten experiments, 3 slices per experiment) + S.E.M. a, percentage of value of fresh slices (i.e., slices that were not preincubated with DMSO or cryopreserved) + S.E.M. ∗, values are significantly higher (P < .05) than those obtained with 12% DMSO.

Histomorphology of fresh and cryopreserved liver slices after 4 h in culture (H&E staining).
Fresh slice (not cryopreserved or preincubated) (A); cryopreserved slice preincubated without DMSO (B); cryopreserved slice preincubated with 12% DMSO (C); cryopreserved slice preincubated with 18% DMSO (D).
Influence of Cryopreservation Medium.
The influence of the medium used as cryoprotectant carrier during preincubation and cryopreservation on viability of slices is shown in Fig. 4. Neither UW nor FCS appeared to be a better cryopreservation medium than WME (Fig. 4, A and B).
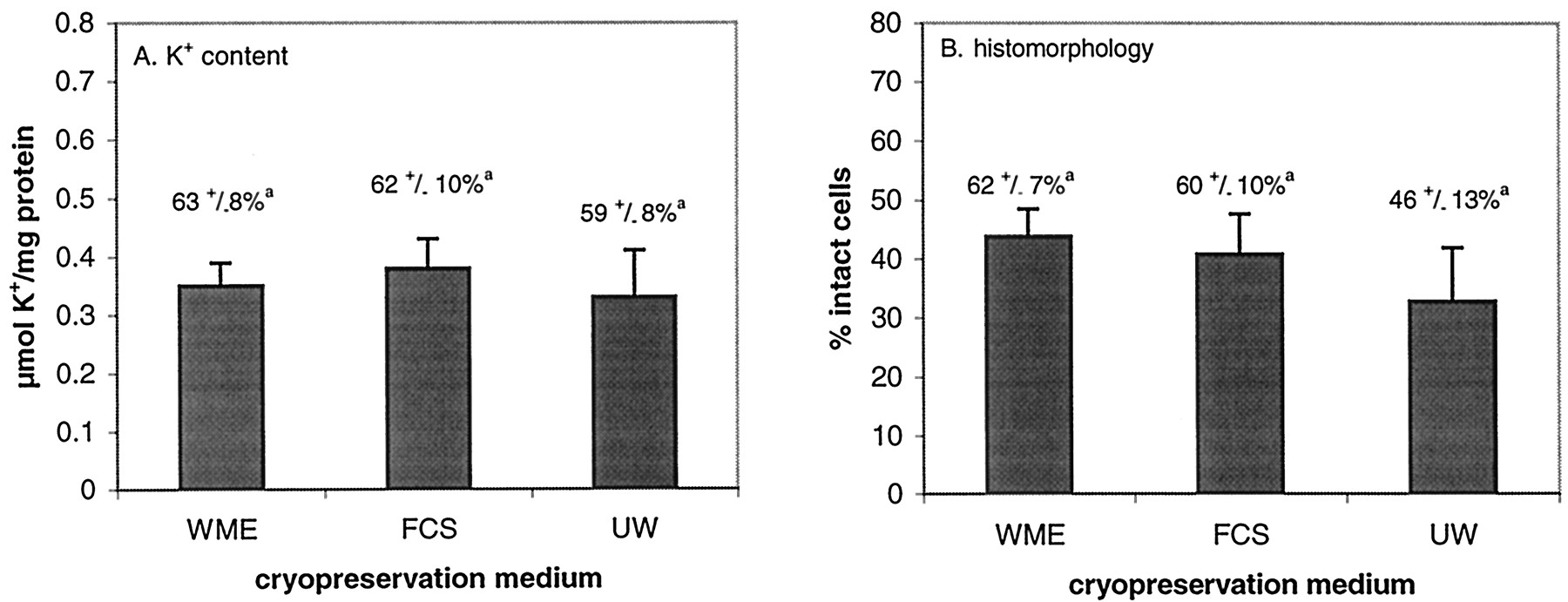
K+ content (A) and histomorphology (% intact cells) (B) in cryopreserved slices preincubated with 18% DMSO in WME, FCS, or UW as preincubation medium.
Slices were cultured for 4 h after thawing. Values are shown as mean of individual experiments (five to seven experiments, 3 slices per experiment) + S.E.M. a, percentage of value of fresh slices (i.e., slices that were not preincubated with DMSO or cryopreserved) + S.E.M.
The Role of Slice Thickness.
When slices of various thickness were prepared, the results after cryopreservation varied (Fig. 5). The results between these experiments were variable, and therefore the results from each separate experiment is given. This shows that the trend in the experiments is consistent and statistically significant; both K+ content and the number of intact cells were considerably decreased in thicker slices. K+retention of the cryopreserved slices reached an optimum in slices with approximately 10 cell layers. Histomorphological examination of slices revealed that damaged cells in cryopreserved slices that are relatively thick, are mainly located at the slice edges and in the cell layers central in the slice. In the thin slice, only the outer cell layers are damaged.

Influence of slice thickness on K+ content (A) and histomorphology (percentage of intact cells) (B) in fresh slices.
Open symbols, fresh slices that were not preincubated with DMSO or cryopreserved; closed symbols, cryopreserved slices. Analogous symbol forms represent fresh and cryopreserved slices from the same experiments. Slices were cultured for 4 h. Values are shown as mean of values of individual slices within the experiments (6 slices per experiment) + S.E.M. Statistically significant results: K+ content of both fresh and cryopreserved slices is statistically significantly related to slice thickness (P < .05). The percentage of intact cells as observed by histomorphological examination is only for cryopreserved slices statistically significantly related to slice thickness.
K+ retention in fresh slices was also related significantly with slice thickness (Fig. 5A), although less marked than in cryopreserved slices. Slice thickness did not affect the number of intact cells in fresh slices (Fig. 5B).
The Role of Cold Storage before Cryopreservation.
Liver tissue and slices are generally kept on ice for approximately 60 min (counting from liver excision) during slice preparation before the cryopreservation preincubation is started. As shown in Fig.6, varying this time from 15 to 75 min, decreased the number of intact cells and K+content, although not statistically significant.

Influence of storage time at 4°C before warm incubation (fresh slices) or cryopreservation (cryopreserved slices, preincubated with 18% DMSO in WME) on K+ content (A) and histomorphology (percentage of intact cells) (B).
Slices are cultured for 4 h. Values are shown as mean values of individual experiments (five to six experiments, 3 slices per experiment) + S.E.M. a, percentage of value of fresh slices (i.e., slices that were stored at 4°C during the same amount of time, but were not preincubated with DMSO or cryopreserved) + S.E.M.
Biotransformation in Liver Slices with Optimized Viability.
In thin slices, cryopreserved according to the improved method (using 18% DMSO in WME), biotransformation activity was determined. Results are shown in Table 1. Formation of 2α-hydroxytestosterone, an important hydroxy metabolite of testosterone in rat slices and a measure for cytochrome P450-mediated phase I biotransformation, was maintained at 60 to 85% of levels of fresh slices. Peak areas of other metabolites (6β-OH, 19-OH, 14α-OH, 16α-OH, 16β-OH, and androstenodione) were maintained at similar levels, as was the total area of all metabolite peaks. Also phase II biotransformation, that is the glucuronidation and sulfation of 7-HC, was maintained at 50 to 90% of fresh values, with a relative constant fraction formed of each of the two metabolites. The biotransformation capacity of cryopreserved slices seems to correlate well with K+ content and slice histomorphology within the same experiment.
Viability and biotransformation capacity of fresh and cryopreserved liver slices after 4 h in culture
Discussion
In an attempt to improve an existing cryopreservation method for liver slices, we found that preincubation with 18% DMSO in WME and direct immersion in liquid nitrogen maintained cellular integrity and functionality of thin slices (8–10 cell layers) at high levels. Since the potassium content, the number of intact cells, and the functionality/drug metabolism parameters are all correlated, the intact cells can be tentatively indicated as viable.
In earlier studies in our laboratory (de Kanter and Koster, 1995) and by others (Glöckner et al., 1996), high viablity was found after fast-freezing with lower concentrations of DMSO (10–12%). However, the viability parameters (i.e., testosterone oxidation, urea synthesis, and retention of ALT) used by de Kanter and Koster (1995) to optimize this cryopreservation method were found to be relatively insensitive to cryopreservation damage comparing them with the parameters that indicate cell integrity like retention of small molecules (GSH, ATP) and potassium and preservation of intact histomorphology (Maas et al., 2000). To prolong incubation with thawed slices, it is important to maintain particularly these sensitive parameters at high levels. Since the biological machinery of the cell is required for the complete expression of cryopreservation damage, slices in the present study were cultured at 37°C for 4 h, before viability testing. Apparently, 18% DMSO maintains sensitive viability parameters better than 12%, during a 4-h incubation period.
As shown by other authors, oxidative drug metabolism (i.e., testosterone oxidation) is well maintained by cryopreservation (Wishnies et al., 1991; Ekins, 1996; Ekins et al., 1996; Glöckner et al., 1996; de Kanter et al., 1998; Maas et al., 2000). However, phase II metabolism is more sensitive to cryopreservation damage (Wishnies et al., 1991; Ekins, 1996; Ekins et al., 1996; de Kanter et al., 1998; Maas et al., 2000). GSH and sulfate conjugation are more sensitive to cryopreservation damage than other metabolic pathways (Steinberg et al., 1999). However, in the present study sulfation was maintained at a high level. There is evidence that conjugation is impaired by cofactor leakage rather than by enzyme inactivation (Maas et al., 2000). The preservation of sulfation in the present study presumably indicates, therefore, that small molecules like the cofactor for sulfation, phosphoadenosine phosphosulfate, are adequately maintained.
In the present study, we selected K+ and histomorphology as endpoints to improve cryopreservation, since we found previously that these parameters not only are very sensitive to cryopreservation damage but also correlate well with cofactor (ATP/GSH) levels in the slice (Maas et al., 2000) and thus we expected these parameters to be reliable predictors of biotransformation activity. As expected, the maintenance of cellular integrity coincided well with the preservation of oxidation and conjugation capacity of cryopreserved slices: 2α-OH testosterone, 7-HC-glucuronidation, and 7-HC-sulfation were produced at levels close to that of fresh slices. Moreover, production of all metabolites was maintained at similar levels, indicating that all metabolic routes of testosterone and hydroxycoumarin were maintained.
The mechanism by which the present procedure protects cells from damage remains a subject of speculation: the IIF might be (largely) prevented by the higher DMSO concentration or the ice crystals may remain smaller. Moreover, DMSO may render the cellular membranes less susceptible for ice crystals formed. It is known that high and toxic concentrations of DMSO (>40%) are required to completely prevent ice formation in aqueous solutions (Boutron and Kaufmann, 1978; Baudot et al., 1999). Therefore, with 18% DMSO the cryopreservation mediumsurrounding the slices will not vitrify during freezing. An even higher concentration, 30% DMSO, did not further increase viability after cryopreservation, probably because of its toxicity.
Other attempts to stimulate vitrification, for example replacing WME by other cryopreservation media (FCS and UW) that contain substances that can behave as cryoprotectant (BSA and hydroxyethyl starch, respectively) did not improve viability. Since these substances remain extracellular, they will only influence extracellular vitrification, which, with this method does not seem to have an added value.
It should be noticed that viability varied within experiments and even more so between the experiments, in contrast to fresh slices. This could be caused by differences between slices derived from different parts of the liver and between livers from different animals. Moreover, the starting conditions of the slices may be critical. We found, for example, that storage time before cryopreservation influences slice viability after cryopreservation.
It can be concluded from the present study that the cryopreservation method improved here, using thin slices preincubated in WME with 18% DMSO as cryoprotectant, offers a simple way to store liver slices and can maintain their viability and biotransformation activity close to that of fresh slices. Variability between slices cryopreserved from different livers may be of concern, but for in vitro metabolism studies, slices from successful cryopreservation experiments can be easily selected by measuring K+ content of a few cryopreserved slices of that particular liver. Because both phase I and II metabolism are maintained quantitatively comparable to fresh slice levels, and more importantly, because even in less successful cryopreservation experiments, metabolite patterns are similar to that of fresh slices, cryopreserved slices seem to be a promising tool to study in vitro metabolism both qualitatively as (semi-) quantitatively.
Footnotes
-
Send reprint requests to: I. A. M. de Graaf, Drug Safety Department, Solvay Pharmaceuticals BV, P.O. Box 900, 1380 DA Weesp, The Netherlands. E-mail: inge.degraaf{at}solvay.com
-
This study was supported by a grant from the Dutch Platform for Alternatives for Animal experimentation.
-
↵2 Theoretically, it is expected that freezing and thawing of slices determine viability after cryopreservation and not the storage period in liquid nitrogen (Mazur, 1984). We compared viability of slices stored for 30 min with slices from the same liver that were stored for approximately 48 h. Indeed, no differences were found.
- Abbreviations used are::
- DMSO
- dimethylsulfoxide
- IIF
- intracellular ice-crystal formation
- UW
- University of Wisconsin medium
- WME
- Williams medium E
- FCS
- fetal calf serum
- Received February 2, 2000.
- Accepted May 11, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}