


Abstract
Human UDP-glucuronosyltransferases (UGT, EC 2.4.1.17) involved in the biotransformation of pyrene were investigated by a sensitive fluorometric high-performance liquid chromatography (HPLC)method developed for determining activities toward 1-hydroxypyrene. The endpoint metabolite of pyrene, 1-pyrenylglucuronide, is a well-known urinary biomarker for the assessment of human exposure to polycyclic aromatic hydrocarbons. 1-Pyrenylglucuronide was synthesized using rat liver microsomes as biocatalyst. The yield was satisfactory, 22%. 1-Pyrenylglucuronide, identified by 1H NMR and by electrospray mass spectrometry, was used for method validation and calibration. The HPLC assay was very sensitive with a quantitation limit of 3 pg (8 fmol) for 1-pyrenylglucuronide. The assay was precise, showing a relative standard deviation of 5% or less at 0.1 to 300 μM 1-hydroxypyrene. Only 2 μg of microsomal protein was required for the assay in human liver. The glucuronidation of 1-hydroxypyrene was catalyzed at high rates in microsomes from pooled or three individual liver samples, showing comparable apparentKm values. The formation of 1-pyrenylglucuronide was catalyzed by recombinant human UGT1A6, UGT1A7, and UGT1A9, the Km values being 45, 12, and 1 μM, respectively. The apparent Km values in human liver microsomes, ranging from 6.9 to 8.6 μM, agreed well with these results. The method provides a sensitive tool for measuring extremely low UGT activities and a specific means for assessing interindividual differences in 1-hydroxypyrene-metabolizing UGT activities in human liver and other tissues.
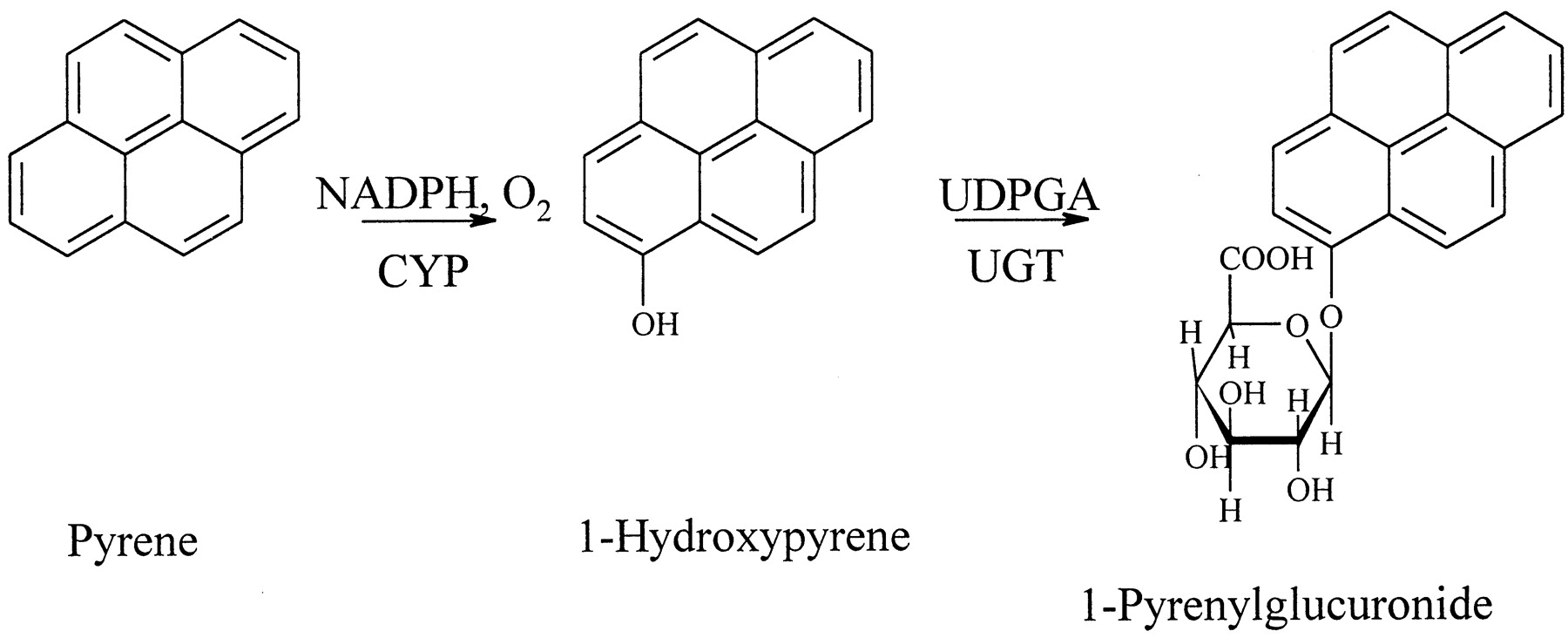
1-Hydroxypyrene (1-HP2) and its glucuronide conjugate are widely accepted as urinary biomarkers of recent polycyclic aromatic hydrocarbon (PAH) exposure (Strickland et al., 1996; Jongeneelen, 1997;Dor et al., 1999). The parent compound pyrene occurs at relatively high concentrations in PAH mixtures (Carmichael et al., 1990; Nylund et al., 1992; Elovaara et al., 1995). Pyrene is metabolized almost exclusively to 1-HP by hepatic cytochrome P450 monooxygenases before it is conjugated and excreted in human urine as a glucuronide conjugate (Strickland et al., 1994). In view of the current knowledge on pyrene exposure, metabolism (Fig. 1), and elimination in humans, it is evident that 1-HP is a substrate for PAH-metabolizing UDP-glucuronosyltransferases (UGT) in vivo. For studying human UGT activities in vitro, we have further developed the method by which 1-HP glucuronidation was assayed in rat liver (Luukkanen et al., 1997).

Metabolism of pyrene in humans.
Enzyme activities in human and rodent tissues have been determined toward compounds that act as substrates for several isoforms of UGT. Such compounds include 1-naphthol (Mackenzie and Hänninen, 1980), 4-methylumbelliferone (Lilienblum et al., 1982), and 4-nitrophenol (Marniemi and Hänninen, 1973; Lautala et al., 1996). Although the glucuronides of these commonly used aglycones are commercially available today, in most methods the lack of glucuronides precludes the direct identification and quantitation of the products. Determination of UGT activities by coupling the glucuronidation reaction to conversion of NADH to NAD+ is a universal method under conditions in which the aglycon substrate does not affect the coupling reaction (Mulder and van Doorn, 1975). The use of14C-labeled UDP-glucuronic acid (UDPGA) with HPLC (Coughtrie et al., 1986; Ethell et al., 1998) or thin-layer chromatography (Bansal and Gessner, 1980) allows the selective quantitation of the radiolabeled glucuronides regardless of the aglycon moiety. Thin-layer chromatography has the disadvantage of not distinguishing conjugates of substrates with multiple glucuronidation sites. In human lungs (Luukkanen et al., 1995), placentas (Paakki et al., 2000), and other extrahepatic tissues such as lymphocytes (Gessner et al., 1978), the specific activities may be several orders of magnitude lower than in the liver. The limited availability of sensitive in vitro methods for the determination of PAH-metabolizing UGT activities in human tissues is a plausible explanation for our limited understanding of the role of human UGTs in the detoxification and elimination of carcinogenic and noncarcinogenic PAH compounds.
This study was undertaken to develop and validate a high-sensitivity assay of human 1-HP UGT activities in vitro using liquid chromatographic quantitation of the reaction product 1-pyrenylglucuronide (HPGA). The reaction was characterized by identification of three UGT isoforms involved in the glucuronidation of 1-HP and by determination of the apparentKm and Vmaxvalues for human liver microsomes and human recombinant UGT1A6, UGT1A7, and UGT1A9.
Materials and Methods
Synthesis of HPGA.
HPGA was synthesized from 1-HP. This reaction was catalyzed by microsomes prepared by differential centrifugation from the livers of male Wistar rats pretreated with Aroclor 1254 (500 mg/5 ml of olive oil per kilogram, intraperitoneally, 5 days before killed) as described (Luukkanen et al., 1997). The reaction mixture was incubated at 37°C. It contained 5 mM (312 mg) UDPGA (disodium salt, Roche Molecular Biochemicals, Ingelheim, Germany; or triammonium salt, Sigma Chemical Co., St. Louis, MO), 5 mM (96 mg) d-saccharic acid 1,4-lactone (Sigma Chemical Co.), and 0.55 mg/ml microsomal protein in a total volume of 100 ml. A 6.9-mg portion of 1-HP (Janssen Chimica, Beerse, Belgium, or Sigma Chemical Co.) in 1 ml of dimethyl sulfoxide (DMSO) was added to the reaction mixture every 30 min for 4 h (totaling 9 × 6.9 mg). The reaction mixture was then incubated at 37°C for 2 h, and the reaction was stopped by adding 10 ml of 15% ZnSO4 and 50 ml of acetonitrile. After sonication the precipitated proteins were removed by centrifugation (4000 rpm, 10 min) and filtration (Durapore HV filter, 0.45 μm; Millipore Co., Bedford, MA). Full conversion of 1-HP to HPGA was verified by HPLC analysis. The crude product was purified by flash chromatography (Baker C18) with 50% acetonitrile in H2O as eluent. HPGA was crystallized at room temperature from a 1:10 mixture of acetonitrile and water, filtered, and washed with a small amount of ice-cold water. The product was identified by accurate mass measurement using low-resolution quadrupole electrospray mass spectrometry and high-resolution fast-atom bombardment mass spectrometry (Kostiainen et al., 1997), and by1H NMR spectroscopy. The purity of the product was checked for residual contents of UDP, UDPGA, andd-saccharic acid 1,4-lactone (a β-glucuronidase inhibitor) according to a previously published method (Luukkanen et al., 1999).
UGT Isoforms 1A6, 1A7, and 1A9.
Human UGT1A6 and UGT1A9 were produced in Chinese hamster lung fibroblast (V79) cells using the Semliki Forest virus (SFV) expression system (Forsman et al., 2000). Cells were infected with recombinant SFV-UGT1A6 or SFV-UGT1A9 virus and harvested at 8 to 16 h postinfection. Cells were lysed by means of several successive freeze-thaw cycles, and the total cell lysate was used for activity assays. UGT activity toward 1-HP (20 or 150 μM) was less than 10% in control V79 cells compared with the cells infected with recombinant SFV-UGT1A6 or SFV-UGT1A9 virus. Commercially available human UGT1A7 Baculosomes were microsomes purchased from PanVera Corporation (Madison, WI). Protein concentrations were determined by the method ofBradford (1976).
Human Liver Microsomes.
Pooled human liver microsomes from Human Biologics International (Scottsdale, AZ) were used for method validation. Three human liver samples were obtained from Oulu University Hospital (Oulu, Finland). The Ethics Committee of the Oulu University Medical Faculty approved the collection of surplus human tissues. The thawed liver samples were homogenized in cold 0.15 M KCl, 50 mM Na-K-phosphate buffer, pH 7.4, to obtain a 25% homogenate for the isolation of the microsomal enzyme fraction by differential centrifugation. The liver homogenate was centrifuged at 10,000g for 15 min and the supernatant at 105,000g for 60 min. The microsomal pellet was suspended in the buffer solution, recentrifuged, and finally resuspended by homogenization in the buffer solution now containing 10% glycerol and stored frozen at −70°C until used. Protein concentrations were determined by the method of Lowry et al. (1951).
1-HP UGT Assay for Human Liver.
The 1-HP UGT assays were carried out in 50 mM phosphate buffer, pH 7.4, at 37°C. The reaction mixture contained 3 mM MgCl2, 2.5 mM UDPGA, and 0.1 to 300 μM 1-HP (added in 5 μl of DMSO) in a final volume of 250 μl. 1-HP was incubated with 2.0 μg (or 5.0 μg) of human liver microsomal protein in the presence of 0.1% BSA. The reaction was started with UDPGA followed by incubation for 10 min. The reaction was stopped by adding 50 μl of ice-cold 15% ZnSO4 and 500 μl of acetonitrile. After 10 min at 4°C, the reaction mixture was sonicated (10 min) and centrifuged (14,000 rpm, 5 min). A 10-μl aliquot of the supernatant was injected to a reversed phase C18chromatography column (Spherisorb S3 ODS-2, 3 μm, 150 × 4.6 mm; Waters Co., Milford, MA). Reaction blanks were incubated without UDPGA. The chromatographic system comprised two LC-10AD pumps, an SCL-10AVP system controller, an SIL-10A autoinjector, and an RF-10AXL fluorescence detector, and Class VP 5.021 software (system 1; Shimadzu, Duisburg, Germany) or a Waters 510 HPLC pump, a Waters 717 Plus autosampler (Waters), a RF-535 fluorescence HPLC monitor (Shimadzu), and Millennium v2.15 software (system 2; Waters). The mobile phase (0.9 ml/min) consisted of 60% acetonitrile in 0.5% aqueous acetic acid. Fluorescence excitation and emission wavelengths of 242 and 382 nm, respectively, were optimal for the detection of HPGA. The enzyme assays were identical except for the protein content, which was 2 μg in system 1 and 5 μg in system 2 due to the lower sensitivity of the latter system. The method was calibrated in the ranges of 2.8 to 55.8 nM (system 1) and 32 to 3169 nM (system 2) with HPGA as reference compound. The enzymatic formation of HPGA was linear over an incubation period of 30 min with 5.0 μg of microsomal protein from human liver.
1-HP UGT Assay for UGT Isoforms 1A6, 1A7, or 1A9.
The assays were carried out as described above with the following modifications. The reaction mixture contained 5 mM MgCl2, 4.4 mM UDPGA, and the reaction was started after a 5-min preincubation period at 37°C with the addition of 1-HP in 5 μl of DMSO. The enzymatic formation of HPGA was linear over an incubation period of 60 min with 1.4 μg (UGT1A6), 15 μg (UGT1A7), or 1.1 μg (UGT1A9) of protein from cells expressing the different isoforms of UGT. Reaction blanks were incubated without UDPGA.
Effect of DMSO.
The specific UGT activities toward 1-HP were determined with 0.4, 0.8, 2.0, 4.0, and 10% DMSO and without DMSO. 1-HP (final concentration 50 μM) was added in 50 μl of acetonitrile and the solvent was evaporated to dryness. Then an appropriate volume of DMSO followed by the standard incubation reagents (50 mM phosphate buffer, pH 7.4, 3 mM MgCl2, 2.5 mM UDPGA, 2.0 μg human liver microsomal protein) was added to a final volume of 250 μl.
Method Validation.
The repeatability of the reaction was determined at three different 1-HP concentrations (1, 50, and 300 μM) with 0.1% BSA or without BSA. The relative standard deviation (RSD) with 0.1% BSA (n = 4) was 2.6% or less at these concentration levels (Table 2). The presence of 0.1% BSA improved the repeatability of the reaction possibly due to the low concentration of microsomal protein. BSA may function as a carrier protein for membrane-bound UGTs, thereby stabilizing the membrane environment of the enzyme. Low concentrations of BSA (0.01–0.5%) had no significant effect on the specific UGT activities at 0.1 and 150 μM 1-HP.
Repeatability of the glucuronidation reaction at 1, 50, and 300 μM 1-HP in human liver microsomes
The limit of detection (system 1) for HPGA was 1.6 fmol (0.6 pg) (signal-to-noise ratio >2) and the limit of quantitation was 7.8 fmol (3.1 pg), determined on the basis of RSD <5% for samples spiked with HPGA (n = 4). The recoveries of HPGA from incubated assay samples were (RSD <5%) 103, 104, and 101% at the tested concentration levels of 10, 51, and 254 nM (n = 4), respectively. Hence, the standard solutions used for calibration could be prepared in the mobile phase without any discrepancy in recovery. The calibration curve was linear over the concentration range 2.8 to 55.8 nM (R2 = 0.999). The samples were stable for at least 1 week when stored refrigerated.
Enzyme Kinetics.
The apparent Km (Michaelis constant) andVmax (maximum velocity) were estimated by fitting the initial glucuronidation velocities observed at 0.1 to 300 μM 1-HP concentrations to the Michaelis-Menten equation by a nonlinear least-squares method (Leonora v1.0, Cambridge University Press, Cambridge, UK). Intrinsic clearance was calculated as theVmax/Km ratio.
Results
HPGA.
The conversion of 1-HP to HPGA by rat liver microsome-assisted synthesis was complete but most of the product was lost during purification. The poor solubility of HPGA complicated the purification of the crude product. HPGA could be dissolved only in mixtures of water and organic solvents. Flash chromatography combined with crystallization from a mixture of acetonitrile and water gave pure HPGA in a satisfactory yield, 25 mg (22%). Since no residual UDP, UDPGA, ord-saccharic acid 1,4-lactone was detected and HPLC analysis showed that the product contained only 0.0025% 1-HP, the product was assumed 100% pure when used as a reference compound. The identity of HPGA was verified by 1H NMR spectroscopy (Table1) and by accurate mass spectrometric measurements. Obtained spectra (electrospray mass spectrometry and fast-atom bombardment mass spectrometry) indicated unambiguously the presence of a molecule [M − H]− ion 393.1 consistent with HPGA, data reported previously (Kostiainen et al., 1997).
Proton magnetic resonances (500 MHz) of 1-pyrenylglucuronide (acetone:D2O, 1:1)
1-HP UGT Assay.
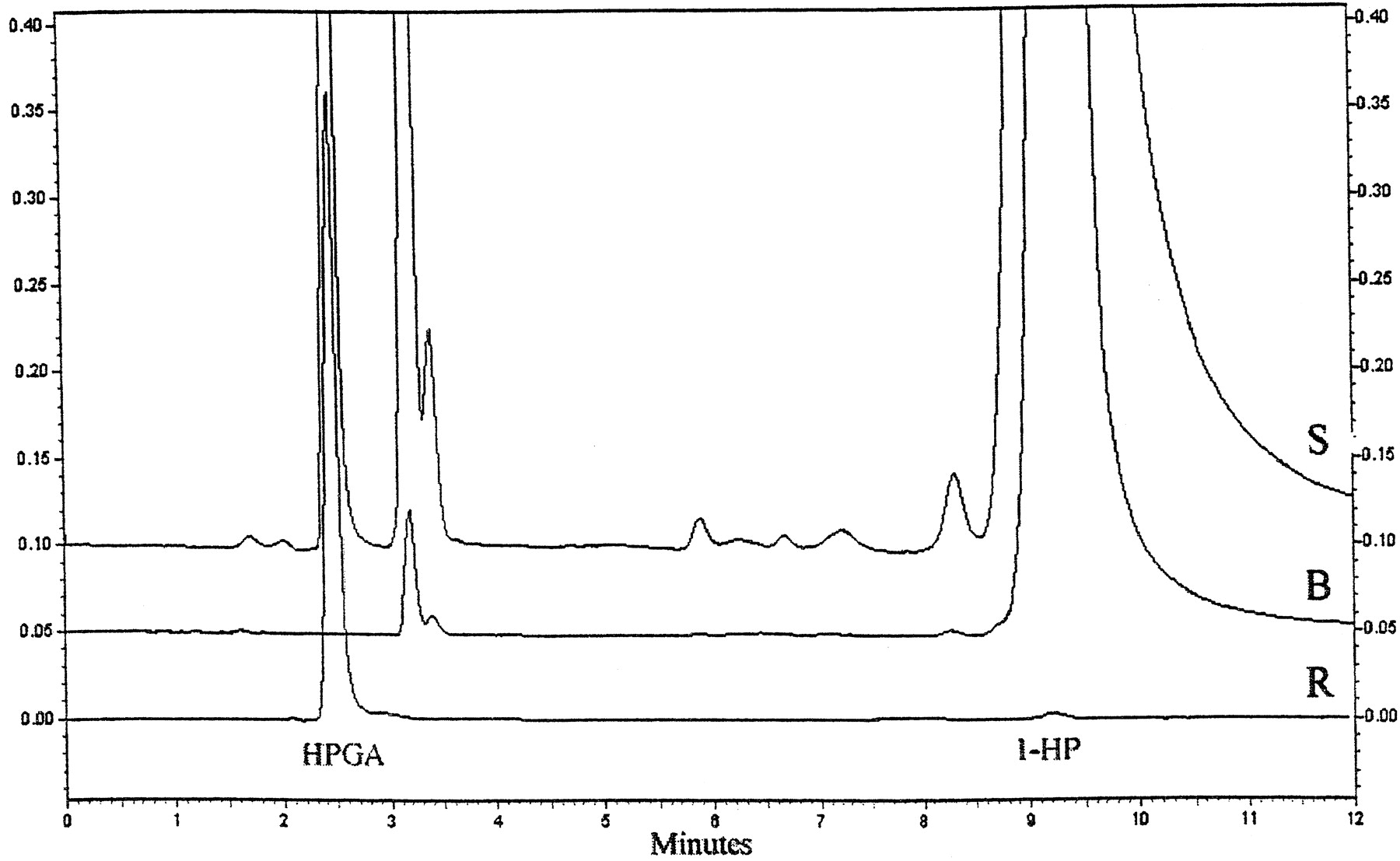
The formation of HPGA was followed by liquid chromatography with fluorescence detection. HPGA eluted at 2.4 min and the aglycon substrate 1-HP at 9.2 min. No interfering peaks at 2.4 min were observed in blank incubations (Fig. 2). This assay has the advantage that it required only protein precipitation and centrifugation but no further preparative steps before chromatographic resolution. Presumably for this reason, the recovery was excellent (approximately 100%) and the precision indicated by the low RSD values was good, too (Table2). The high sensitivity of this assay (quantitation limit 8 fmol) allowed determination of UGT activity at 1-HP concentrations as low as 0.1 μM and upwards. Notably, a 0.1% BSA is recommend for standard assays in view of our results on method validation and optimization (see Materials and Methods).

Typical chromatograms from an assay with 150 μM 1-HP (S), from a blank incubation with 150 μM 1-HP (B) and of a 254 nM HPGA-standard (R).
1-HP was added in DMSO. This vehicle has a detergent-like effect on membrane-bound enzyme activity as revealed by experiments conducted for this purpose at 50 μM 1-HP and at 0, 0.4, 0.8, 2.0, 4.0, and 10% concentrations of DMSO. In these assays, the activity was low in the absence of DMSO, exhibiting approximately only one-eighth of the activity that was detectable under assay conditions with the vehicle. DMSO seemed to have some inhibitory effect on the glucuronidation reaction at test concentration of 10%. Administration of 1-HP in 5 μl of DMSO (2%) was found to be optimal for keeping 1-HP in solution and maintaining maximum UGT activity. Detergents such as Triton X-100 had no additive effect on enzyme activity in incubations containing DMSO.
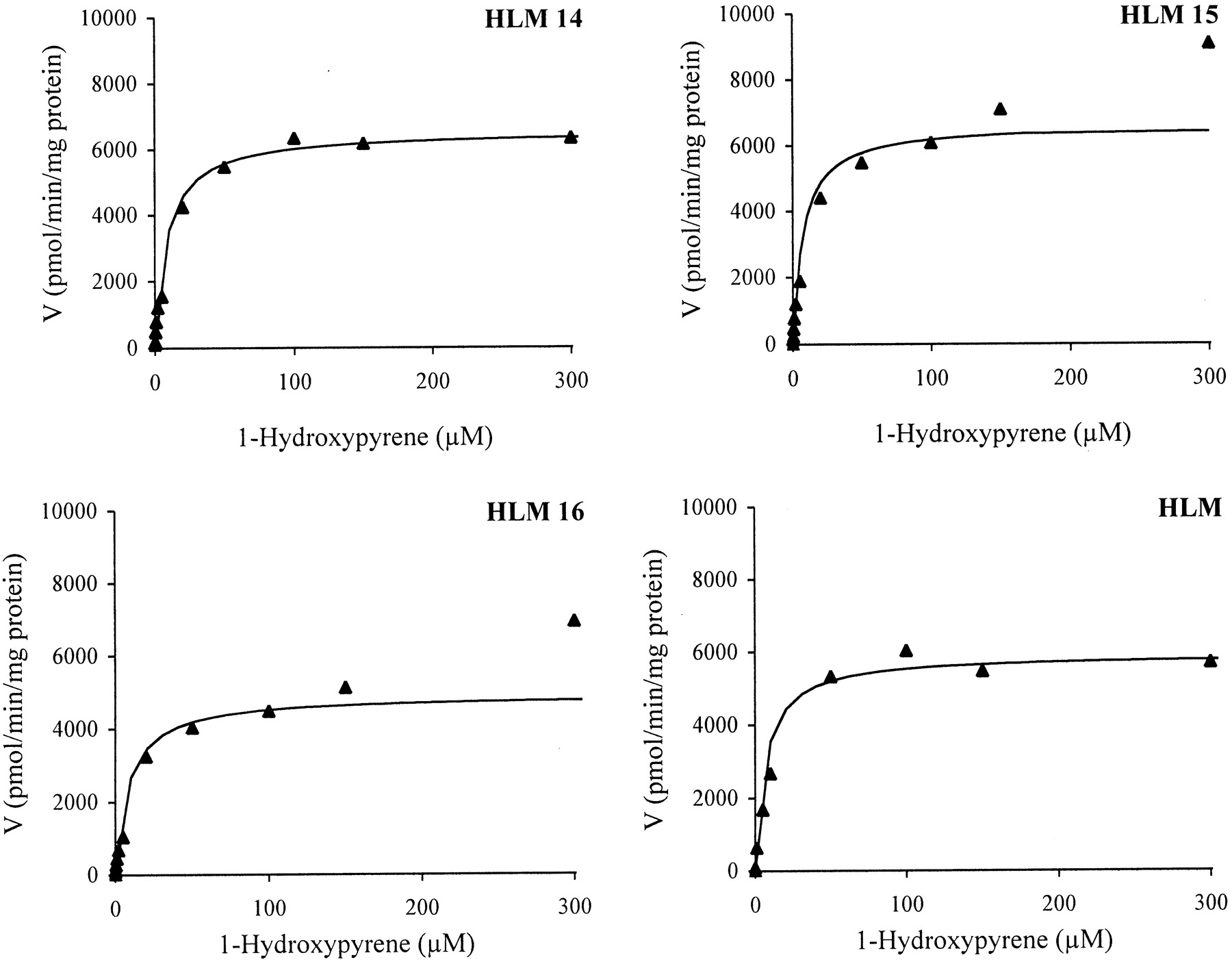
Glucuronidation of 1-HP by Human Liver Microsomes and Human UGT1A6, UGT1A7, and UGT1A9.
Glucuronidation of 1-HP was catalyzed by liver microsomes and by three cDNA-expressed enzymes: UGT1A6, UGT1A7, and UGT1A9. The initial velocities of the glucuronidation reaction were determined at 0.1 to 300 μM 1-HP. Care was taken not to exceed a substrate turnover of 10%. The apparent Km andVmax values (Table3) were estimated for the recombinant UGTs and liver microsomes using the Michaelis-Menten equation for hyperbolic enzyme kinetics. The apparent Kmand Vmax values were essentially the same in liver microsomes from three donors, ranging from 7.4 to 8.6 μM and from 4.9 to 6.6 nmol/min/mg of protein, respectively. Similar values were obtained also with pooled hepatic microsomes as determined in two laboratories and with two different chromatographic systems. The apparent Km andVmax values were 6.95 ± 0.59 μM and 5.84 ± 0.27 nmol/min/mg of protein (system 1, Table 3) and 4.51 ± 0.56 μM and 4.75 ± 0.27 nmol/min/mg of protein (system 2), respectively.
Enzyme kinetic parameters for the glucuronidation of 1-HP in human liver microsomes (HLM) and by human recombinant UGTs
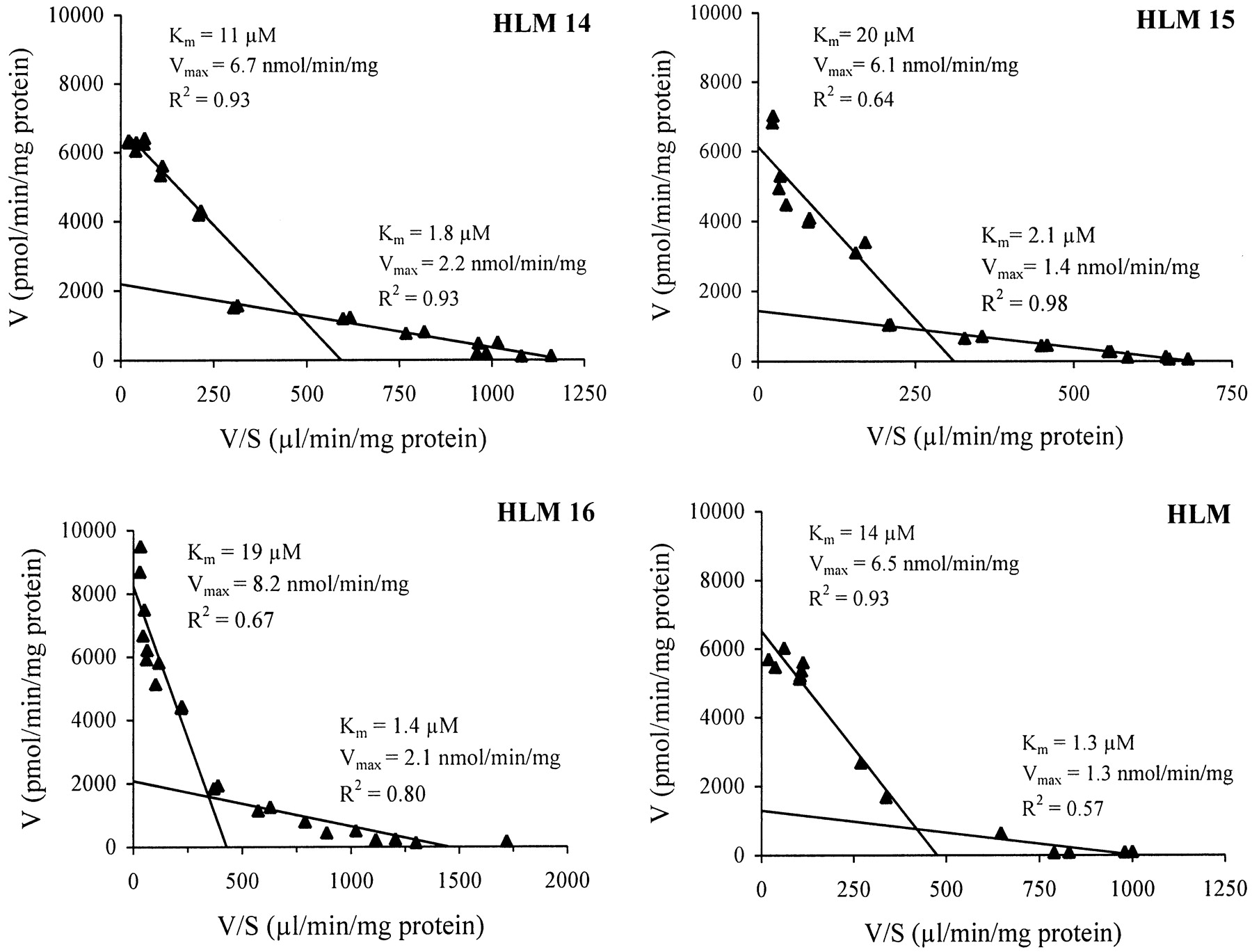
The Michaelis-Menten plots are shown in Fig.3, and the corresponding Eadie-Hofstee plots in Fig. 4. The clearly biphasic nature of the Eadie-Hofstee plots (each yielding two straight lines) suggested that this glucuronidation pathway was catalyzed at least by two hepatically expressed enzymes. The apparent kinetic parameters estimated by the Eadie-Hofstee method gave aKm(1) of 1.3 to 2.1 μM and aKm(2) of 11 to 20 μM, and aVmax(1) of 1.3 to 2.2 nmol/min/mg and aVmax(2) 6.1 to 8.2 nmol/min/mg of protein, indicating contribution by a high-affinity and a low-affinity component, respectively. The difference in affinity for 1-HP, as implicated by the above-mentioned two Kmvalues, was not marked, however (Fig. 4).

Enzyme kinetics of 1-HP UGT in three individual human liver samples (HLM 14–16) and pooled human liver microsomes (HLM).
Each data point is the mean of at least two determinations.
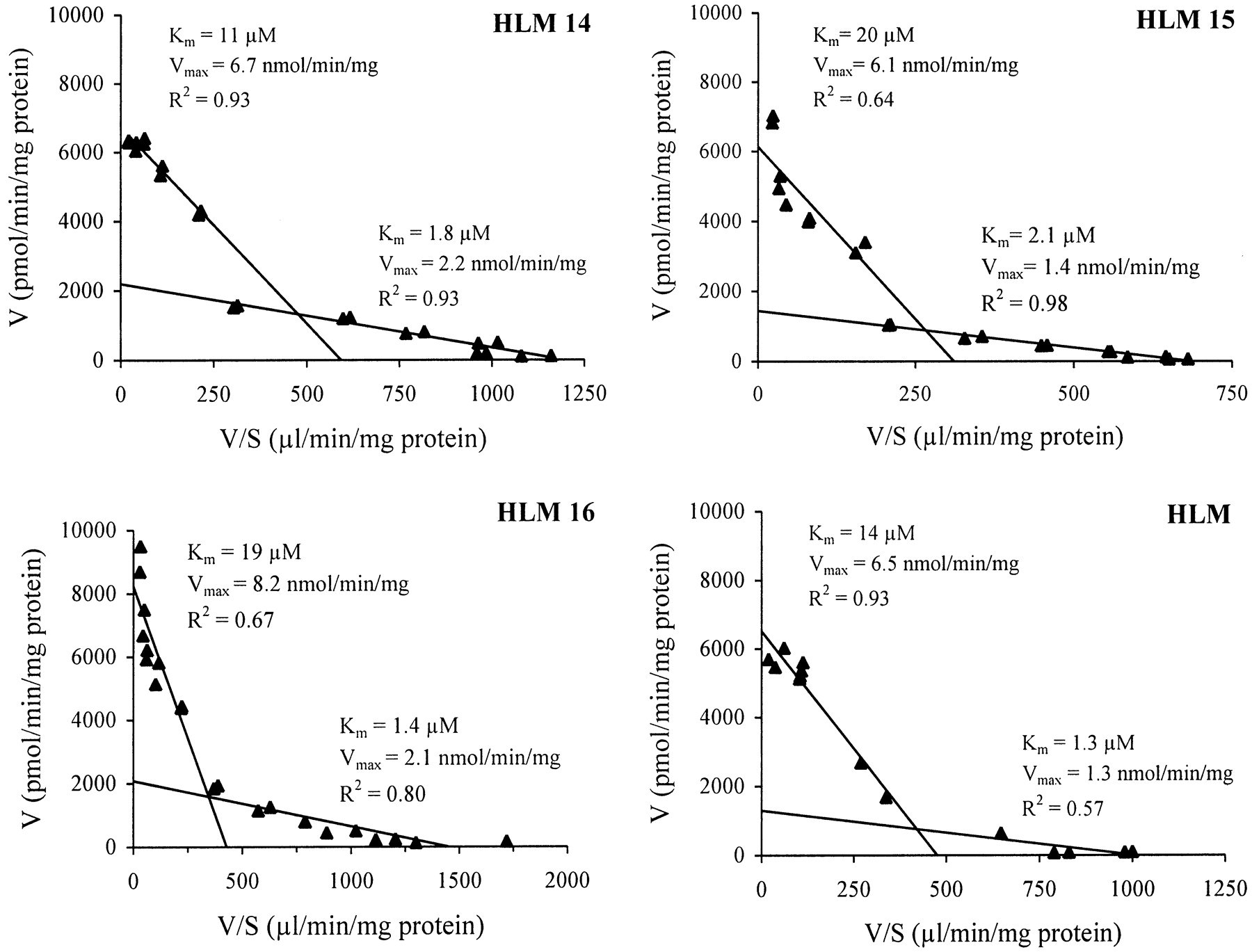
Eadie-Hofstee plots of 1-HP UGT in three individual human liver samples (HLM 14–16) and pooled human liver microsomes (HLM).
The glucuronidation of 1-HP by recombinant UGT enzymes was consistent with monophasic Michaelis-Menten kinetics (plots not shown). All three isoforms exhibited relatively low apparentKm values (1–45 μM), variation inVmax values (0.15–0.57 nmol/min/mg), and large variation in intrinsic clearance (3–568 μl/min/mg). UGT1A9 appeared to be the most effective isoform in conjugating 1-HP.
Discussion
A sensitive fluorometric HPLC method was developed by which 1-HP glucuronidation activity and kinetics were readily assayed in human liver microsomes as well as in cells expressing the recombinant human UGTs 1A6, 1A7, and 1A9. The quantitation limit of the 1-HP UGT method (8 fmol) was several orders of magnitude lower than the quantitation limit (0.1 nmol) reported for HPLC methods using14C-labeled UDPGA for product detection (Coughtrie et al., 1986; Ethell et al., 1998). Thus, the present method allowed detection even of extremely low UGT activities in human liver and other tissues. In this context, the activities quantitated in 36 human placentas provide a noteworthy example for the applicability of this method (Paakki et al., 2000).
Michaelis-Menten enzyme kinetics was determined under initial reaction rate conditions, starting with an aglycon concentration as low as 0.1 μM 1-HP. Characterization of the glucuronidation reaction revealed that the velocity data obtained for the recombinant UGTs showed a Michaelian behavior consistent with a kinetically single enzyme. The apparent Km value for UGT1A9 (1 μM) was conspicuously low in comparison with those obtained for UGT1A6 (45 μM) and the extrahepatic isoform, UGT1A7 (12 μM) (Table3). According to our results, even low UGT activity can be accurately measured in human liver by a routine procedure using only 2 μg of microsomal protein, 1-μM 1-HP concentration, and a 10-min incubation.
The apparent Km values observed for the glucuronidation of 1-HP were lower than those reported, e.g., for the glucuronidation of 6-hydroxychrysene by the human isoforms UGT1A6 or UGT1A9 (Bock et al., 1993, 1998). The lowKm values for the glucuronidation of 1-HP (Km = 1 μM) and 6-hydroxychrysene (Km = 7–20 μM) by human UGT1A9 indicate the importance of this form in conjugating PAH compounds at low tissue concentrations. The Km values for the glucuronidation of 1-HP (Km = 45 μM) and 6-hydroxychrysene (Km = 20–140 μM) by human UGT1A6 are also relatively low. Evidently, UGT1A6 and UGT1A9 play a key role in conjugating PAH phenols in human liver. The lowKm for the glucuronidation of 1-HP by UGT1A7 suggests that this extrahepatic isoform is involved in conjugation of PAH phenols.
UGT1A9 yielded the highestVmax/Km value for the glucuronidation of 1-HP, making it the most efficient catalyst among the human isoforms (Table 3). It should be noted that theVmax values shown in Table 3 for the human recombinant UGT1A6, UGT1A7, and UGT1A9 forms may not be directly comparable because their expression levels were not known. UGT1A9 may therefore not be the only major form responsible for the glucuronidation of 1-HP in human liver. This conclusion is based on the results obtained for hepatic microsomes, which showed an apparentKm value (6.9–8.6 μM) between theKm values of the hepatic enzymes UGT1A9 (1 μM) and UGT1A6 (45 μM). The Eadie-Hofstee plots, which were clearly biphasic in all liver samples, provided further evidence in this line.
In view of the present results, 1-HP may be a sensitive marker substrate for PAH conjugating UGTs, i.e., UGT1A9 and other low-Km isoforms. The same isoforms of human recombinant UGTs (1A6, 1A7, 1A9) that we found active toward 1-HP have previously been shown to be active toward many other PAH compounds of concern to human health. For instance, UGT1A6 catalyzes the glucuronidation of benzo(a)pyrene-3,6-quinol, 3,6-dihydroxychrysene (Gschaidmeier et al., 1995) and 4-, 5-, 8-, and 12-hydroxybenzo(a)pyrenes (Jin et al., 1993). UGT1A7 catalyzes the glucuronidation of 7-hydroxybenzo(a)pyrene (Strassburg et al., 1999b). UGT1A9 catalyzes the glucuronidation of a wide variety of bulky phenolic compounds, including hydroxylated PAHs, such as benzo(a)pyrene-3,6-quinol (Gschaidmeier et al., 1995), 6-hydroxychrysene (Bock et al., 1993), and 7-hydroxybenzo(a)pyrene (Strassburg et al., 1999b). UGT1A8 and UGT1A10 glucuronidate several hydroxylated metabolites of benzo(a)pyrene and 2-acetylaminofluorene (Mojarrabi and Mackenzie, 1998). Notably, UGT1A6 and UGT1A9 are expressed in both hepatic and extrahepatic tissues, whereas UGT1A7, UGT1A8, and UGT1A10 are extrahepatic forms (Strassburg et al., 1997, 1999a; Mojarrabi and Mackenzie, 1998). Within the UGT2 family, UGT2B7 is a human form capable of glucuronidating hydroxylated metabolites of benzo(a)pyrene and 2-acetylaminofluorene (Jin et al., 1993).
Human UDP-glucuronosyltransferases are known to catalyze the glucuronidation and elimination of hydroxylated metabolites of various PAH compounds, thereby decreasing their toxicity, e.g., carcinogenicity and teratogenicity. The mutagenicity of PAH compounds stems from the formation of DNA-reactive metabolites and ultimate carcinogens such as benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide. The genoprotective role of UGTs in the metabolism of benzo(a)pyrene has been demonstrated in animal studies (Hu and Wells, 1994; Vienneau et al., 1995; Kim and Wells, 1996). The reduction in the mutagenicity of benzo(a)pyrene and benzo(a)pyrene-3,6-quinone in the Ames test upon addition of UDPGA further emphasizes the role of glucuronidation as a true detoxification route (Bock et al., 1990). The level of UGT-mediated metabolism of PAH phenols is therefore an important determinant of cell susceptibility to PAH-mediated toxicity.
In conclusion, 1-HP is a large-molecular PAH readily glucuronidated by human liver microsomes as well as by the human recombinant isoforms UGT1A6, UGT1A7, and UGT1A9. Unlike benzo(a)pyrene, pyrene has not been recognized as a human carcinogen (IARC, 1983). It is tempting to speculate that 1-HP is rapidly eliminated and detoxified as a result of the high rates (Table 3) of glucuronidation at even low 1-HP concentrations in human liver. Our findings may explain the lack of carcinogenicity reported for pyrene, which is the parent PAH compound of mammalian pathways producing 1-pyrenylglucuronide in vivo.
Acknowledgments
We gratefully acknowledge Professor Olavi Pelkonen for helpful criticism and Professor Harri Vainio for support during the early phases of this work. We also thank Ulla Peltonen for skillful technical assistance.
Footnotes
-
↵1 Present address: Viikki Drug Discovery Technology Center, Department of Pharmacy, P.O. Box 56 (Viikinkaari 5E), FIN-00014 University of Helsinki, Finland.
-
This work was supported by the Commission of the European Communities, Biomed 2 program (BMH4-CT97-2621).
- Abbreviations used are::
- 1-HP
- 1-hydroxypyrene
- PAH
- polycyclic aromatic hydrocarbon
- UGT
- UDP-glucuronosyltransferase
- UDPGA
- UDP-glucuronic acid
- HPGA
- 1-pyrenylglucuronide
- DMSO
- dimethyl sulfoxide
- BSA
- bovine serum albumin
- HPLC
- high-performance liquid chromatography
- SFV
- Semliki Forest virus
- RSD
- relative standard deviation
- Received January 9, 2001.
- Accepted April 26, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}