


Abstract
The CYP3A subfamily enzymes are the most abundant and important drug-metabolizing enzymes. Wide variation in the CYP3A5 expression was well known. Recently, G−44 to A of CYP3AP1was found to segregate with CYP3A5*3 defective allele. The homozygous A−44 subjects showed low expression of CYP3A5. In Caucasian, only 9.2% of CYP3AP1 alleles were with G−44 and associated with the wild-type CYP3A5*1 allele, which expressed CYP3A5 significantly. By using polymerase chain reaction and FauI endonuclease digestion, we found that 28% of CYP3AP1 alleles are G−44in 110 Chinese subjects. The frequency is 3 times higher in Chinese than in Caucasian, implying more Chinese subjects are probably extensive CYP3A5 metabolizers. In two Chinese subjects, we also found a heterozygous G13048gt-to-G13048gc mutation at the intron 5 splicing donor site, leading to a splicing defect. A 6478-base pair minigene, including intron 4 to intron 7, was used for in vitro transcription. Both the wild-type and the mutated minigenes produced splicing variants. The wild-type minigene used Ggt13050 as the splicing donor. The mutant minigene used gt8504 in intron 4 or gt13112 in intron 5 as the splicing donor for various splicing acceptors. The splicing defect may result in a shorter peptide or cause the frame shift. In the other two Chinese subjects, we found A14763-to-G mutation in exon 7, resulting in the Q200R amino acid change. The consequence of the polymorphism site has not been known. In Caucasian, there is a reported T398N polymorphism. In these Chinese subjects, we did not find polymorphism at this site.
The CYP3A enzymes are the most important subfamily of cytochrome P450s in xenobiotic metabolism. Four CYP3A genes have been described in humans:CYP3A4, CYP3A5, CYP3A7, andCYP3A43 (Hashimoto et al., 1993; Gellner et al., 2001). Among them, CYP3A7 is a fetal protein. CYP3A4 and CYP3A5 are functional enzymes in adults and metabolize a wide range of xenobiotics. The sequences of CYP3A4 and CYP3A5 genes are available recently. In 2000, we published the CYP3A4sequence in the GenBank database (accession no. AF209389). We have also aligned CYP3A5 cDNA sequence with the human genome databaseAC005020. The 15810 to 47610 bp1 of AC005020 was assigned as CYP3A5 gene, comprising the transcriptional initiation site (Jounaidi et al., 1994) to exon 13. In this study, we renumbered the position 15810 of AC005020 as position 1. The cDNA sequences of CYP3A4 and CYP3A5 have been characterized with 90% similarity. Interestingly, similarity is not only at the coding region; the intron 5 of CYP3A4 andCYP3A5 also show 90% identity. In a genetic polymorphism study, the intron sequence is usually used to design specific primers to amplify exon fragments by PCR. It was difficult to design specific primers to study the genetic polymorphism of CYP3A enzymes. This difficulty explains the late discovery of CYP3A4 and CYP3A5 polymorphism in the attempt of finding interindividual variation for major cytochrome P450s.
Overlapping substrate specificity between CYP3A4 and CYP3A5 has also made it difficult to separate the metabolism of these two enzymes. Although a wide interindividual variation of CYP3A metabolism has been known, little phenotypic data have been produced to reveal variation in CYP3A5 activity in humans. However, there is evidence for wide variation in the expression of CYP3A5. Both immunoblotting and Northern blot analysis have detected CYP3A5 expression in only 10 to 30% of human livers (Aoyama et al., 1989; Wrighton et al., 1990; Schuetz et al., 1994). Previously, a point mutation (10%) of Thr398Asn (CYP3A5*2) was found (Jounaidi et al., 1996). It was postulated that the amino acid change caused protein instability and the low level of CYP3A5 expression. The postulation was, however, not well evidenced. Paulussen et al. (2000) found two linked mutations, A/G−45and T/G−369. The mutations were well associated with the CYP3A5 expression. Recent publications indicate that A/G−45 polymorphism identified by Paulussen et al. (2000) is in fact the A/G−44 polymorphism in the promoter of the pseudogene CYP3AP1 (Finta and Zaphiropoulos, 2000; Gellner et al., 2001). Furthermore, Kuehl et al. (2001) found a complete concordance between A/G−44 polymorphism and CYP3A5*3 defect allele in Caucasians. Only the subjects with G−44 inCYP3AP1 had normal CYP3A5 expression.
In this study, we examine the genetic polymorphism in Chinese by SSCP analysis. We found a novel missense mutation, an intron mutation causing splicing defect, and an ethnic difference in the polymorphism at the reported sites in CYP3AP1. Furthermore, we also demonstrated that the splicing defect at intron 5 could result in many splicing variants by in vitro transcription.
Materials and Methods
Genomic DNA Isolation.
Blood samples were obtained from 75 healthy unrelated subjects and 35 stroke patients from Chinese (Han) population living in Taiwan. The samples were from two previous studies of CYP2D6 and CYP3A4 polymorphism. DNA was isolated from peripheral leukocytes using a DNA isolation kit (Puregene; Genta System Inc., Minneapolis, MN).
PCR-SSCP and Sequencing Analysis.
Exons (including the exon-intron boundaries) and part of the 5′ upstream region from −438 to +105 bp were amplified by PCR in separate reactions. The primer sets and PCR conditions used are shown in Table1. The PCR reaction was carried out in 50 μl of solution consisting of 5 μl of 10× Taq buffer, 0.2 μM dNTPs, 0.06 to 0.3 μM of each primer, 0.1 μg of genomic DNA as template, and 2.5 U of Taq polymerase (TaKaRa, Kyoto, Japan). To carry out SSCP gel electrophoresis, a GeneGel Excel 12.5/24 kit (Amersham Pharmacia Biotech, Uppsala, Sweden) was used. The PCR product (2.5–4.5 μl) was mixed with an equal volume of denatured dye, heated to 95°C for 10 min, cooled on ice for 5 min, and then spun at 4°C. Five microliters of heat-denatured PCR product was directly applied onto the gel. The gel was electrophoresed on a GenePhor unit (Amersham Pharmacia Biotech) at 600 V, 25 mA, 15 W and kept at suitable running temperature. After a 2-h electrophoresis, the gel was transferred into a Hoefer automatic gel stainer (Amersham Pharmacia Biotech) according to the manufacturer's instructions. PCR products showing differences on SSCP analysis were subjected to direct sequencing using ABI PRISM Big Dye terminator cycle sequencing ready reaction kit and ABI PRISM 377-96 DNA sequencer (Applied Biosystems, San Francisco, CA).
Primers and PCR conditions for PCR-SSCP, PCR-RFLP, and others
PCR-RFLP for Gln200Arg (CYP3A5*4) in Exon 7.
A PCR-based test of Gln200Arg was developed. The A14763 → G change in the sequenceAAGAC creates the recognition sited of BsmAI (GAGAC). The PCR reaction was carried out in 50 μl of solution consisting of 5 μl of 10× Taq buffer, 0.2 μM dNTPs, 0.06 μM primer EX7(S) and EX7(R) (Table 1), 0.1 μg of genomic DNA as template, and 2.5 U ofTaq polymerase. After PCR amplification, the DNA fragments were digested with BsmAI before electrophoresis using a 12.5% polyacrylamide gel. Samples with A14763gave 317-bp band, while samples with G14763 gave 149- and 168-bp bands (Fig. 1).

Detection of polymorphism by PCR-RFLP.
Following PCR and digestion with the appropriate enzyme, oligonucleotides were analyzed by gel electrophoresis. A, detection of A/G14763 by BsmAI digestion. Lanes 1 and 2 [without (−) and with (+) BsmAI digestion] are from a sample of homozygous A14763. Lanes 3 to 7 [without (−) and with (+) BsmAI digestion] are from samples of heterozygous A/G14763. B, detection of G13048gt → gc by Hsp92II digestion. Lanes 1 and 2 [without (−) and with (+) Hsp92 II digestion] are from a sample of homozygous G13048gt. Lanes 3 to 6 [without (−) and with (+) Hsp92 II digestion] are from samples of heterozygous G13048gt/c.
PCR-RFLP for G13048gt → gc Splicing Defect (CYP3A5*5) in 5′ End of Intron 5.
A PCR-based test of G13048gt → gc was developed. The G13048gt → gc change in the sequence TATG creates the recognition site ofHsp92II (CATG). The PCR reaction was carried out in 50 μl of solution consisting of 5 μl of 10× Taqbuffer, 0.2 μM dNTPs, 0.3 μM primer EX5(S) and EX5(R) (Table 1), 0.1 μg of genomic DNA as template, and 2.5 U of Taq polymerase. After PCR amplification, the DNA fragments were digested with Hsp92II before electrophoresis using a 12.5% polyacrylamide gel. Samples with G13048gt gave 4- and 248-bp bands, while samples with G13048gc gave 4-, 81-, and 167-bp bands (Fig. 1).
CYP3A5 Minigene Construction.
Genomic DNA samples of G13048gt → gc splicing defect were amplified by Extra-Long PCR (GeneAmp XL PCR kit; Applied Biosystem, Branchburg, NJ). The PCR reaction was carried out in 25 μl of solution consisting of 7.5 μl of 3.3× XL buffer, 0.1 μM dNTPs, 0.16 μM minigene sense primer and minigene reverse primer (Table 1), 1.1 mM Mg(OAc)2, 0.1 μg of genomic DNA as template, and 1 U of γTth DNA polymerase (Applied Biosystem). The PCR fragments were amplified from genomic DNA of sample with heterozygous G13048gt → gc splicing defect. The minigenes (the wild-type and the mutant type) added with 3′-polyA were cloned into TA cloning kit (Invitrogen, San Diego, CA) and subcloned into the eukaryotic expression vector pcDNA3.1V5/His/TOPO (Invitrogen) with endonucleases KpnI and NotI (Fig.2). The nucleotide sequence in the recombinant plasmids containing the 5′ end of intron 4 to 3′ end of intron 7 of CYP3A5 gene was confirmed.
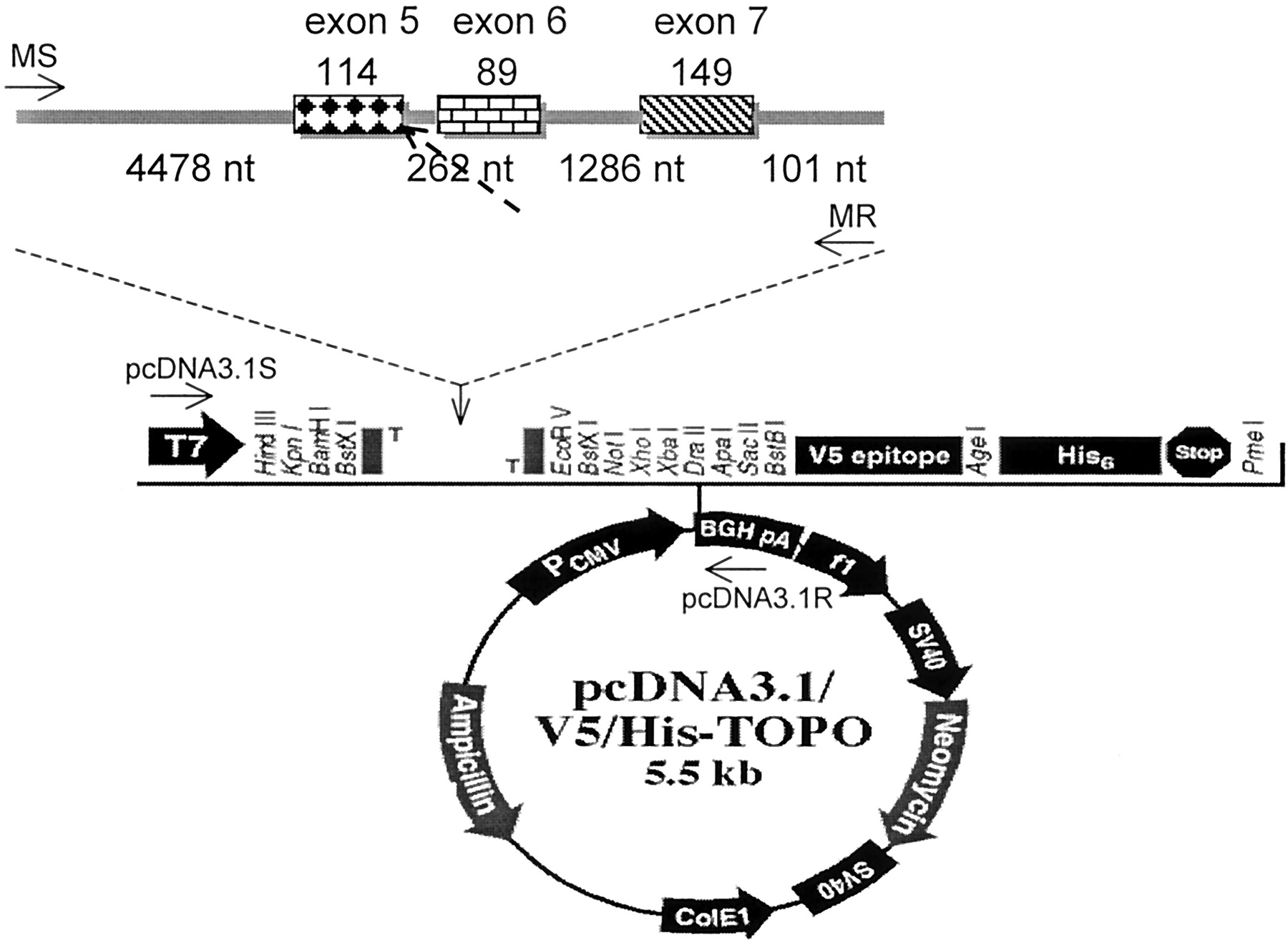
Location of PCR primers and map of expression vector containing CYP3A5 minigene.
Analysis of Splicing of Pre-RNA Transcribed from Minigene.
The minigene constructs were transfected as LipofectAMINE complexes into Caco-2 cells. After 4.5 h, the transfection medium was removed and fresh medium was added. Cells were grown for another 36 h before harvesting. Total RNA was isolated by use of Ultraspec-II RNA isolation system (Promega, Madison, WI) and reverse transcription PCR (RT-PCR) was performed. To characterize the expression of the mRNA from the minigen that contained the splice-donor site mutation, we used the two pcDNA 3.1 sense (pcDNA 3.1S) and reverse (pcDNA 3.1R) primers to perform the PCR reaction, using a GeneAmp PCR kit (Table 1 and Fig. 2). Aliquots from cells transfected with the vector alone, wild-type, and mutated minigenes were analyzed by RT-PCR and electrophoresis on 1.5% agarose gel. The multiple splicing variants from the wild-type and mutated minigenes were further subcloned and the PCR products were sequenced. The primers located at CYP3A5 cDNA XL-4kb(S) and XL-4kb(R) (Table 1) were used to amplify the endogenously expressed CYP3A5 at Caco-2 cells as the internal control.
PCR-RFLP Detection Assay for the G−44 → A Mutation in CYP3AP1.
All PCR assays were performed using a 1 in 50 dilution of the original 3A51F/3A52R PCR product as template. PCR conditions used are shown in Table 1. For G−44 → A mutation, the PCR reactions was carried out in 50 μl of solution consisting of 5 μl of 10× Taq buffer, 0.2 μM dNTPs, 0.3 μM primer A−44G(S) and A−44G(R) (Table 1), and 2 U of γTaq polymerase. A PCR-based test of A−44G was developed. The G−44 → A mutation changed the recognition site of FauI (CCCGC) into the sequence CCCAC. After PCR amplification, the DNA fragments were digested with FauI before electrophoresis using a 2.5% agarose gel. Samples with A−44 gave 335-bp band, while samples with G−44 gave 147- and 188-bp bands (Fig. 3).

FauI analysis of the CYP3A5 A−44G mutation site on 2.5% agarose gel.
Lanes 2 and 3 show positive control [without (−) and with (+)FauI digestion] and lanes 4 and 5 show homozygous A−44; lanes 6 and 7 show heterozygous A−44G; lanes 8 and 9 show homozygous G−44; and lane 1 shows 1-kb DNA ladder.
Results
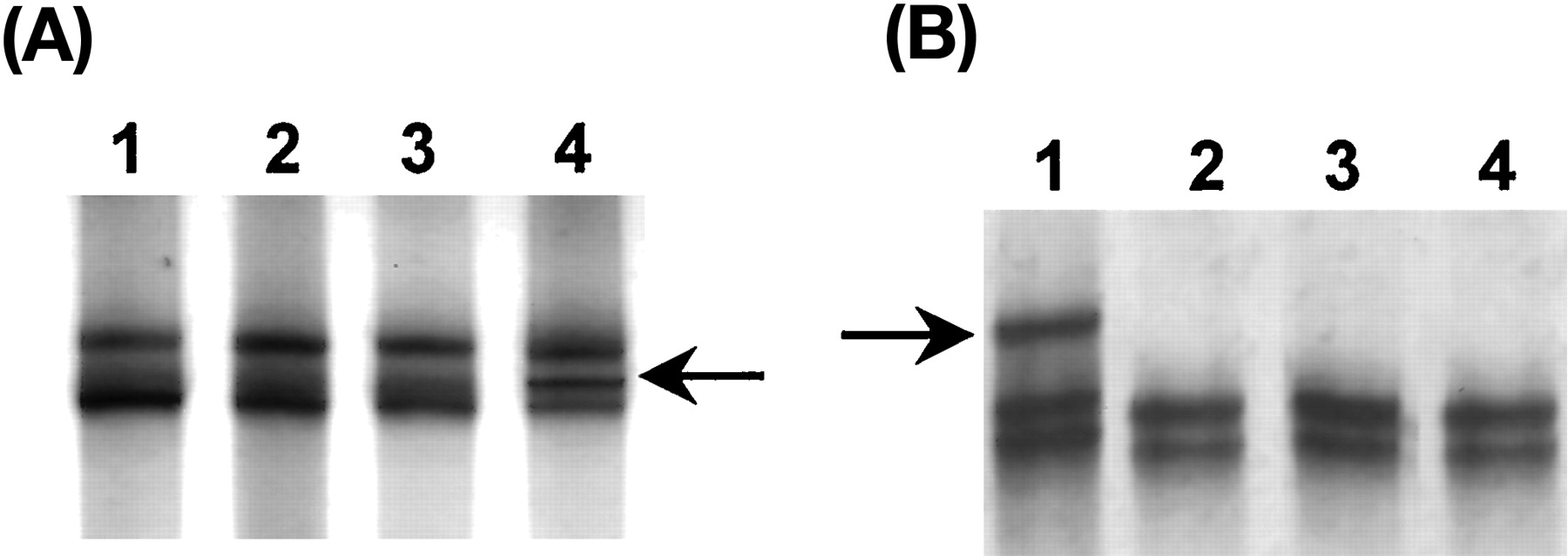
In addition to A−44G polymorphism ofCYP3AP1, we have screened exons 4, 5, 6, 7, 8, 10, 11, and 12 of CYP3A5 for possible genetic polymorphism by SSCP (Fig.4). Two mutations were found (Table2). One is Q200R (CYP3A5*4) and the other is at intron 5 splicing donor site (CYP3A5*5). The T398N mutation (CYP3A5*2) reported in the literature was not found in this study. By using PCR-RFLP with appropriate endonuclease (Fig. 1), the incidence rate of each mutation was found to be 1% (two heterozygous subjects among 110 subjects, 2 of 220). The two rare mutations do not explain the wide interindividual variation of CYP3A5 expression. The association of A−44G polymorphism with defective CYP3A5*3 allele is particularly interesting. The samples were screened with FauI endonuclease (Fig. 3). The percentage of G−44 allele in Chinese is much higher than in white subjects (Table 3).

Detection of polymorphisms in exon 7 and intron 5 by SSCP analysis.
A, exon 7, lane 4 shows the sample of heterozygous A/G14763, whereas lanes 1 to 3 show the samples of homozygous A14763 sample. The arrow indicates the single-stranded band of altered migration. B, intron 5, lane 1 shows the sample of heterozygous 5′ end splicing donor site of intron 5 G15048gt → gc, whereas lanes 2 to 4 show the samples of G15048gt.
Polymorphic sites of CYP3AP1 and CYP3A5 in Chinese
Comparison of A/G−44 polymorphism between white and Chinese subjects
A 6478-bp minigene (nt 8458–14935) containing intron 4, exon 5, intron 5, exon 6, intron 6, exon 7, and intron 7 was inserted into an expression vector pcDNA 3.1 (Fig. 2). The mRNA produced by in vitro transcription was detected by RT-PCR (Fig.5). The endogenous CYP3A5 expression in Caco-2 cells was also amplified as a positive control using primers from exon 5 to exon 9 and gave a 503-bp band. After in vitro transcription, the pcDNA3.1 vector produced a 257-nt cDNA fragment. After inserting the wild-type or mutated minigenes, the cryptic splice sites are at gu8504 in intron 4 and ag14925 in intron 7. The wild-type minigene containing G13048gt produced two cDNA bands by RT-PCR in Caco-2 cells. One is a 664-bp product (the 352-nt exon 5, 6, 7, the 257-nt vector, the first 45 nt from the intron 4 before the cryptic site, and the last 10 nt from the intron 7 after the cryptic site), and the other is 575-bp product (89-bp exon 6 less). The mutated minigene containing G13048gc produced multiple splicing products in Caco-2 cells (Fig. 5). The cDNA sequence indicated that one of them used the second gt13009 in intron 5 as the splicing donor for intron 5. The size of intron 5 became 200 nt. The PCR product was 726 bp, which contains the additional 62-nt intron 5 fragment. The splicing variant is out of the open reading frame. The other three transcripts all used the second gt8504 in intron 4 to replace gt13050 as the splicing donor site. The second product is to splice gu8504 with ag13310 of intron 5, resulting the cDNA product without exon 5 (550 bp; 104 nt less, in-frame product). The third product used the ag14685 of intron 6 to delete exon 5 and 6 and gave a 461-bp product (203 nt deleted, out of frame). The last product is to splice using the ag14925of intron 7 to give the product containing no exon (312 bp; 352 nt less, out of frame). They represent various possibilities to use alternative splicing donor and acceptor sites (Fig.6). Regardless, the G13048gc produced defective mRNA, which may produce a shorter peptide or out of reading frame.

RT-PCR amplification of mRNA from Caco-2 cells transfected with minigene constructs.
Lane 3, amplicon from pcDNA3.1/V5/His/TOPO vector alone as positive control (257 bp). Lane 4, amplicon of mRNA expressed from the Caco-2 cells without transfection. Lane 5, amplicon of mRNA expressed from the pcDNA3.1/V5/His/TOPO vector. Lane 6, amplicon of mRNA expressed from the wild-type CYP3A5 minigene. Lane 7, amplicon of mRNA expressed from the mutant type CYP3A5 minigene. Lanes 8 and 9, amplicon of mRNA expressed from the CYP3A5 endogenous as internal control (503 bp). Lane 1, 1 kb+ DNA ladder. Wild-type mRNA yielded 664- and 575-bp bands, whereas the mutant-type mRNA yielded 312-, 461-, 550-, and 726-bp bands.

Predicted splicing mechanism using cryptic sites.
Discussion
The CYP3A5*3 allele is probably the most important mutant allele of CYP3A5. Kuehl et al. (2001) showed that A/G−44 polymorphism in CYP3AP1 is linked to the splicing defect of CYP3A5*3. Subjects with G−44 in CYP3AP1 have CYP3A5 protein expression in Western blot analysis, but not for the A−44 subjects (CYP3A5*3). Accounting for both homozygous G−44 and heterozygous A−44G subjects, there will be 54 of 300 in white subjects (Paulussen et al., 2000) and 57 of 110 in Chinese with CYP3A5 expression. The percentage in Chinese (52%) who might have CYP3A5 expression is much higher than the percentage in white subjects (18%). Because of overlapping substrate specificity of CYP3A4 and CYP3A5, this discrepancy has not been documented in the literature. Few substrates can be used to delineate CYP3A4 and CYP3A5 metabolism. Midazolam can be a probe drug (Gorski et al., 1994; Haehner et al., 1996). CYP3A5 metabolizes midazolam preferably through 1-hydroxylation rather than 4-hydroxylation. The level of CYP3A4 expression in Chinese is still unknown. Although the average clearance of midazolam is not necessarily higher in Chinese, the average ratio of 1-hydroxy-midazolam to midazolam or 4-hydroxy-midazolam (CYP3A4 metabolite) can be expected to be higher in Chinese.
Jounaidi et al. (1994) sequenced two human CYP3A5 clones with different 5′-flanking sequences. Clone 1 contained A−44and T−369, and clone 2 contained G−44, G−369, and a large number of other nucleotide changes. The A/G−44was assigned as A/G−45 by Paulussen et al. (2000), while the T/G−369 was the same. It is now clear that these sequences are not from the promoter ofCYP3A5, but from the pseudogene CYP3AP1 (Finta and Zaphiropoulos, 2000; Gellner et al., 2001). It was postulated that the A/G−44 and T/G−369polymorphism is derived from the two clones (Paulussen et al., 2000). We have sequenced the region from five subjects with homozygous A−44 and five subjects with homozygous G−44. They all showed corresponding linked −369 mutation, but their sequence is identical with that of clone 1. None of the 10 subjects showed sequence of clone 2. Apparently, A/G−44 and T/G−369 sites are polymorphic sites of CYP3AP1, not the PCR product from the other CYP3A locus sequence.
Jounaidi et al. (1996) reported a T398N mutation in 2 of 38 alleles in Caucasian. In this study, we found none in 110 subjects. Instead, we found a point mutation of Q200R in 2 of 220 alleles. The percentage of this mutation in other ethnic groups is still unknown. It was postulated the T389N is associated with CYP3A5 expression level. The postulation remains to be verified. The effect of both T398N and Q200R mutation on CYP3A5 activity is to be studied by site-directed mutagenesis.
In this study, we also found 2 of 220 alleles showed a mutation at the intron 5 splicing donor site. We have prepared a minigene containing nearby exons and introns (Fig. 2). In vitro transcription has shown various splicing variants. The shift of splicing donor and acceptor sites created different mRNAs. Some of them are out of reading frame. For example, the introduction of 62-nt intron 5 fragment into mRNA sequence will cause a frame shift and create an early stop codon. One possible mRNA product is 114 nt shorter but is still in the open reading frame. The in vitro transcription revealed different possibilities; which product represents the splicing of CYP3A5*5 in vivo is unclear. The mutation is also worth studying in other ethnic groups to explain the wide interindividual variation of CYP3A activities.
Footnotes
-
This work was supported by Grant NHRI-GT-EX89S831L from the National Health Research Institute of Republic of China (Taipei).
- Abbreviations used are::
- bp
- base pair
- PCR
- polymerase chain reaction
- SSCP
- single-strand conformational polymorphism
- RFLP
- restriction fragment length polymorphism
- RT-PCR
- reverse transcription-polymerase chain reaction
- nt
- nucleotide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}