


Abstract
Metabolism of the prototype human CYP2D6 substrates debrisoquine and bufuralol proceeds at a much slower rate in mice; therefore, the mouse has been proposed as an animal model for the human CYP2D6 genetic deficiency. To interpret the molecular mechanism of this deficiency, a cDNA belonging to the CYP2D gene subfamily (Cyp2d22) has been cloned and sequenced from a mouse mammary tumor-derived cell line. In the current study, Cyp2d22 enzyme was overexpressed and purified from insect cells using a baculovirus-mediated system. The activity of this purified enzyme was directly compared with purified human CYP2D6 toward codeine, dextromethorphan, and methadone as substrates. Purified Cyp2d22 was found to catalyze the O-demethylation of dextromethorphan with significantly higher Km values (250 μM) than that (4.2 μM) exhibited by purified human CYP2D6. The Km for dextromethorphan N-demethylation by Cyp2d22 was found to be 418 μM, much lower than that observed with human CYP2D6 and near the Km for dextromethorphan N-demethylation catalyzed by CYP3A4. CYP2D6 catalyzed codeine O-demethylation, whereas Cyp2d22 and CYP3A4 mediated codeine N-demethylation. Furthermore, methadone, a known CYP3A4 substrate and CYP2D6 inhibitor, was N-demethylated by Cyp2d22 with a Km of 517 μM and Vmax of 4.9 pmol/pmol/min. Quinidine and ketoconazole, potent inhibitors to CYP2D6 and CYP3A4, respectively, did not show strong inhibition toward Cyp2d22-mediated dextromethorphan O- or N-demethylation. These results suggest that mouse Cyp2d22 has its own substrate specificity beyond CYP2D6-like-deficient activity.
The superfamily of cytochrome P450 (P450) genes encodes a large number of structurally similar, heme-containing monooxygenases, which are found in virtually all living organisms, from bacteria to yeast, fungi, plants, mammals, and humans. In mammals and humans, P450 enzymes are mostly expressed in hepatic tissues and catalyze the oxidative metabolism of a wide variety of xenobiotics (Ortiz de Montellano, 1995). Many P450 isoforms are also expressed in extrahepatic tissues, such as the skin, lung, kidney, breast, nasal mucosa, and brain (Peters et al., 1989; Amet et al., 1997; Gilham et al., 1997; Hedlund et al., 1998; Wolkenstein et al., 1998; Voirol et al., 2000; Siegle et al., 2001). These are increasingly understood to play an important role in the biosynthesis and biodegradation of endogenous compounds in addition to the biotransformation of xenobiotics, an area of active investigation in many laboratories.
CYP2D6 is a polymorphic human P450 isoform involved in the metabolism of about 25% of all prescribed drugs, including antiarrhythmics, antihypertensives, β-blockers, antipsychotics, and tricyclic antidepressants (Gonzalez et al., 1988; Skoda et al., 1988; Gonzalez and Idle, 1994; Nebert, 1997). CYP2D6 has also been shown to metabolize carcinogens and neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (Fonne et al., 1987; Gilham et al., 1997; Nebert, 1997), 1,2,3,4-tetrahydroquinoline (Ohta et al., 1990), and indolealkylamines (Yu et al., 2003). A functional deficit of CYP2D6 activity is observed in up to 10% of Caucasians, who are termed poor metabolizers compared with extensive metabolizers with normal activity. In addition to the pharmacokinetic clinical implications, poor metabolizers appear more likely to develop neurodegenerative disorders such as Alzheimer's or Parkinson's disease (Saitoh et al., 1995; McCann et al., 1997). Mouse and dark agouti strain rat have been proposed as animal models for this human CYP2D6 genetic deficiency because they also have an impaired ability to metabolize CYP2D6 probe drugs such as debrisoquine and bufuralol (Al-Dabbagh et al., 1981; Kahn et al., 1985; Gonzalez et al., 1987; Masubuchi et al., 1997), although some discrepancy is recognized from enzyme kinetics and gene analysis (Matsunaga et al., 1989; Adams et al., 1991; Barham et al., 1994). A Cyp2d enzyme partially purified from mouse liver microsomes does not exhibit debrisoquine 4-hydroxylase activity (Masubuchi et al., 1997). Recently, a cDNA belonging to the Cyp2d gene subfamily, termed Cyp2d22, was cloned and sequenced from a mouse mammary tumor-derived cell line (Blume et al., 2000). This Cyp2d22 enzyme has an identical amino acid sequence as that of the partially purified Cyp2d enzyme sequenced at the N terminus (Masubuchi et al., 1997). Immunoblot analysis of mouse tissues with highly specific anti-Cyp2d22 antisera indicates that Cyp2d22 protein is most abundant in mouse liver and also present in adrenal, ovary, and mammary glands at intermediate levels (Blume et al., 2000).
The aim of the current study was to understand the enzymatic function of Cyp2d22 isoform and to contribute to the interpretation of the molecular mechanism for CYP2D6 deficiency phenotype in mouse species. The Cyp2d22 enzyme was overexpressed in insect cells using a baculovirus-mediated system, purified with affinity chromatography, and its whole molecular mass was determined by electrospray ionization/liquid chromatography mass spectrometry (ESI/LC-MS) analysis. Kinetic analyses with multiple substrates and inhibitors were performed for recombinant Cyp2d22 and compared with human CYP2D6 and CYP3A4. The results indicated that this murine Cyp2d22 enzyme, although possessing a deficit of CYP2D6-like substrate specificity, has its intrinsic substrate specificity, and enzymatic activities are rather similar to human CYP3A4.
Materials and Methods
Chemicals. Dextromethorphan, dextrorphan, 3-methoxymorphinan, 3-hydroxymorphinan, fluoxetine, and norfluoxetine standard samples were purchased from Research Biochemicals International (Natick, MA). Codeine, norcodeine, morphine, methadone, 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP), 2-mercaptoethanol, imidazole, reduced NADPH, l-α-dilauroylphosphatidylcholine, phenylmethylsulfonyl fluoride (PMSF), and 60% perchloric acid were purchased from Sigma (St. Louis, MO). Testosterone and 6β-testosterone were from Steraloids (Newport, RI). 3-[(3-Cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate was bought from Pierce (Rockford, IL). Ni-NTA Superflow was purchased from Qiagen (Valencia, CA). Emulgen 911 was from Kao-Atlas (Tokyo, Japan). High-performance liquid chromatography (HPLC) solvents and other chemicals were of the highest grade commercially available and were used as received.
Molecular Biology. CYP3A4 enzyme along with P450 reductase and cytochrome b5, expressed in BTI-TN-5B1-4 insect cells using baculovirus system, and the control insect cells infected with wild-type virus were purchased from BD Gentest (Woburn, MA). Restriction enzymes were bought from Roche Diagnostics Corporation (Indianapolis, IN), Invitrogen (Carlsbad, CA), or New England Biolabs (Ipswich, MA) and were used in buffer systems provided by the manufacturers. High-five Trichoplusia ni cells were obtained from Invitrogen. HyQCCM-3 media and fetal bovine serum were from Hy-Clone Laboratories (Logan, UT). General molecular biology methods were performed using standard procedures (Sambrook et al., 1989), and routine insect cell culture methods were followed as described by O'Reilly et al. (1994).
Cyp2d22 and CYP2D6 cDNAs were subcloned into pIZT/V5-His vector to introduce an N-terminal hexahistidine tag. DNA sequences were confirmed by complete gene sequencing using an Applied Biosystems ABI-Prism 377 and Perkin-Elmer ABI BigDye Terminator cycle sequencing kit. These cDNAs were transferred from pIZT/V5-His into pFastBac/hexahistidine-tagged (HT) vector (Life Technologies) using similar digestion procedures for creation of recombinant baculovirus.
Protein Expression and Purification. Rat NADPH P450 reductase was expressed and purified from bacterial cultures according to published procedures (Shen et al., 1991). Spectral P450 activity was determined by the method of Omura and Sato (1964) and was used as a means to establish optimal conditions for P450 expression and purification. Expression, purification, and biomedical characterization of unmodified CYP2D6 enzyme were reported elsewhere (Yu et al., 2001, 2002). Baculovirus-mediated HT-Cyp2d22 and HT-CYP2D6 were overexpressed in T. ni suspension cultures using a baculovirus expression system as CYP2C9 (Haining et al., 1996; Yu et al., 2001) and purified using affinity column chromatography. All the purification steps were carried out at 4°C, and all the buffers were at pH 7.4. The crude insect cell pellet was homogenized in solubilization buffer [20% (v/v) glycerol, 1 mM PMSF, 1% (v/v) Emulgen 911, and 20 mM 2-mercaptoethanol in 50 mM potassium phosphate buffer] by making 5 to 10 passes with a glass/Teflon homogenizer. P450 isoform was solubilized by stirring the homogenized insect cell pellet for 60 min, and the insoluble material was removed by centrifugation at 100,000g for 60 min. The supernatant was loaded onto a Ni-NTA Superflow column (1 ml/20 nmol of P450) preequilibrated with solubilization buffer at 30 ml/h. After the sample was loaded, the column was washed with four columns of wash buffer A (0.5 M sodium chloride in solubilization buffer) at 30 ml/h. Then the column was washed overnight with wash buffer B [20% (v/v) glycerol, 1 mM PMSF, 2 mM 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate, 0.5 M sodium chloride, 5 mM imidazole, and 20 mM 2-mercaptoethanol in 50 mM potassium phosphate buffer]. The column was further washed with four columns of wash buffer C (25 mM imidazole in wash buffer B), followed by the elution with 100 mM imidazole in wash buffer B. The fractions containing P450 enzyme were combined and dialyzed overnight against buffer D [20% (v/v) glycerol and 0.1 mM EDTA in 100 mM potassium phosphate buffer]. Purified HT P450 isoform was aliquoted and stored at –80°C until further use. Purity is estimated at greater than 95% as estimated by SDS-polyacrylamide gel electrophoresis, followed by staining with Coomassie Blue (Fig. 1).
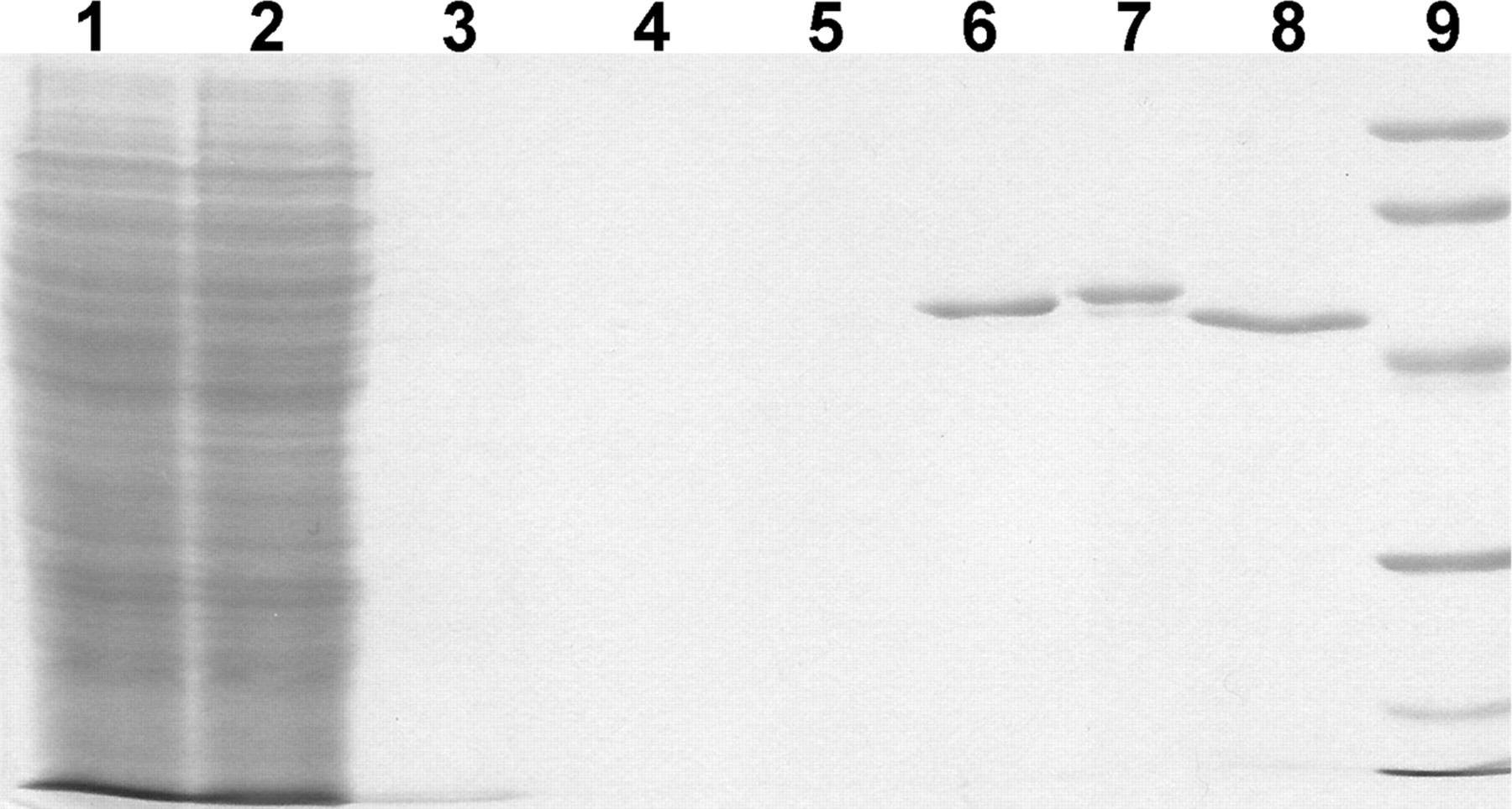
SDS-polyacrylamide gel electrophoresis analysis of various fractions during the purification of HT-Cyp2d22. A resolving gel polymerized from 9% acrylamide was used to separate proteins using a standard Tris borate-EDTA buffer system, followed by staining with Coomassie Blue. Lane 1, membrane preparation from T. ni culture expressing HT-Cyp2d22. Lane 2, flow-through from Ni-NTA Superflow during loading. Lane 3, pooled fractions with wash buffer A. Lane 4, pooled fractions with wash buffer B. Lane 5, pooled fractions with wash buffer C. Lane 6, final purified HT-Cyp2d22. Lane 7, final purified HT-CYP2D6 by following the same protocol. Lane 8, final purified nonpolyhistidine-tagged CYP2D6 (Yu et al., 2001). Lane 9, molecular mass markers in descending order: phosphorylase b, 97.4 kDa; bovine serum albumin, 68.0 kDa; ovalbumin, 43.0 kDa; carbonic anhydrase, 29.0 kDa; and lysozyme, 14.3 kDa.
ESI/LC-MS Analysis of Purified Proteins. ESI/LC-MS analyses were performed on a Micromass Quattro II tandem quadrupole mass spectrometer (Micromass Ltd., Manchester, UK) coupled to an HPLC [Shimadzu LC-10AD with SPD-10AV UV-visible variable detector (Shimadzu Scientific Instruments, Inc., Columbia, MD)]. The instrument was controlled by a computer running Windows NT (Microsoft, Redmond, WA)-based Micromass MassLynxNT 3.2 software. The source temperature was 150°C with the cone voltage set to 55 kV. Solvent flow through the POROS R2 perfusion column (2.1 × 150 mm) from Perseptive Biosystems (Cambridge, MA) was 0.2 ml/min with 100% of the flow (50 pmol of protein injected) being diverted to the mass spectrometer. Acquisition was carried out from m/z 500 to 2000 Da in the continuum scanning mode. ESI mass spectra were collected; the individual scans across the HPLC peak were combined; and each spectrum was deconvoluted using the MaxEnt program.
Assay of Enzymatic Activity. P450 enzymes were reconstituted with rat P450 reductase (1:2 ratio) and dilauroylphosphatidylcholine as described previously (Yu et al., 2001) before the addition of substrate. All the reactions were carried out at 37°C in 100 mM potassium phosphate, pH 7.4, in a final volume of 200 μl. Reactions were optimized for protein and time with respect to linearity before kinetic analysis. Reactions were initiated by the addition of 20 μl of 10 mM NADPH after 5-min preincubation with the substrate at 37°C. For the kinetic analyses, dextromethorphan concentrations in the incubations were 1to50 μM for (non)HT-CYP2D6-mediated O-demethylation, 0.5 to 8 mM for (non)HT-CYP2D6-catalzyed N-demethylation, and 25 to 3000 μM for HT-Cyp2d22- and CYP3A4-catalyzed O- and N-demethylation. Codeine concentrations were 50 to 3000 μM for investigation of its O-demethylation and 0.2 to 8 mM for N-demethylation. Final fluoxetine concentrations in the reactions were 0.5 to 50 μM for (non)HT-CYP2D6 and 10 to 250 μM for HT-Cyp2d22 and CYP3A4. Methadone final concentrations in the incubations were 10 to 1000 μM for examination of the N-demethylation. Testosterone concentration was fixed at 50 or 250 μM to investigate whether Cyp2d22 catalyzes its hydroxylations. Same concentrations of testosterone were used for the control reactions with CYP2D6 and CYP3A4. Negative control incubations for all the studies included reactions without the addition of reductase, P450 isozyme, substrate, or NADPH. For inhibition studies, dextromethorphan concentration was fixed at 1 μM (for O-demethylation) and 1 mM (N-demethylation), and quinidine and ketoconazole concentrations ranged from 5 to 10,000 nM and 10 to 3000 nM, respectively. To determine the Ki value for quinidine against HT-CYP2D6 activity, three different concentrations were used for dextromethorphan (1, 3, and 10 μM), and five different concentrations were used for quinidine (0, 5, 10, 50, and 100 nM). All the reactions were carried out in duplicate. After 5- to 10-min incubation, the reactions were terminated with 10 μl of 70% perchloric acid, except for the reactions with testosterone and methadone, which were stopped with 200 μl of ice-cold acetonitrile. The mixture was vortexed for 20 s, cooled on ice for 10 min, and centrifuged at 14,000g for 10 min. The supernatant was transferred to a new vial and directly injected for HPLC or LC-MS analysis.
HPLC and LC-MS Quantification of Metabolites. HPLC analyses were carried out on a Waters Alliance system consisting of the 2690 separation module, the 2487 dual λ absorbance detector, and the 474 scanning fluorescence detector controlled with Millennium32 software. A 4.6 × 250 mm Spherisob 5 μM phenyl analytical column (Waters, Milford, MA) was used to separate dextromethorphan, codeine, fluoxetine, methadone, and their metabolites. A 150 × 4.6-mm i.d. Nucleosil C18 column (Supelco, Bellefonte, PA) was used to analyze testosterone and its metabolites. Analysis of dextromethorphan and its metabolites was performed essentially as described previously (Yu and Haining, 2001). Dextromethorphan and its metabolites 3-methoxymorphinan and dextrorphan were eluted at 13.6, 10.7, and 8.36 min, respectively. HPLC analyses of fluoxetine and norfluoxetine, codeine and its metabolites, methadone and its metabolites, and testosterone and 6β-hydroxytestosterone were carried out according to these published methods (Venn and Michalkiewicz, 1990; Pierce et al., 1992; Norman et al., 1993; Purdon and Lehman-McKeeman, 1997) with some modifications. Generally, separation of fluoxetine and its metabolites was achieved with a mobile phase containing 60% buffer E (10 mM potassium phosphate monobasic, pH 3.5, adjusted with orthophosphoric acid) and 40% acetonitrile. The excitation and emission wavelengths of the fluorescence detector were set at 235 and 310 nm, respectively. Fluoxetine and its N-demethylated metabolite norfluoxetine were eluted at 14.1 and 11.9 min. The mobile phase consisted of 65% water with 0.1% trifluoroacetic acid and 35% acetonitrile, and a water mixture (400/600, v/v) and was used to separate codeine and its metabolites norcodeine and morphine, which were eluted at 15.8, 9.2, and 8.2 min. The excitation and emission wavelengths of the fluorescence detector were set at 280 and 335 nm, respectively, for the analysis of codeine and its metabolites. Methadone and its metabolites were separated with a mobile phase containing with 30% buffer E and 70% acetonitrile and were detected with the UV detector set at 210 nm. Methadone and EDDP were eluted at 13.6 and 16.7 min, respectively. Gradient elution (50–80% methanol in water from 0 to 10 min) was used to separate testosterone and its metabolites, which were detected using the UV detector with a wavelength set at 240 nm. 6β-Hydroxytestosterone and testosterone were eluted at 5.16 and 8.88 min, respectively.
ESI/LC-MS analyses were used to quantify EDDP produced from methadone, which were performed using a Micromass ZMD 4000 quadrupole mass spectrometer (Waters) coupled to a Waters Alliance HPLC System. The instrument was controlled by a computer running Windows NT-based Micromass MasslynxNT V3.5 software. The optimized instrument parameters were as follows: source temperature, 100°C; cone voltage, 29 kV. Isocratic elution with 30% water containing 0.1% trifluoroacetic acid and 70% acetonitrile was used to separate the metabolite and substrate. Solvent flow through the Spherisob phenyl analytical column was 1.0 ml/min with 35% of the flow diverted to the mass spectrometer. Raw data were collected in the selected-ion recording mode with m/z set to 278 Da.
Data Analysis. Enzyme Michaelis-Menten parameters Km and Vmax were estimated by nonlinear regression (GraphPad Prism 3.02, GraphPad Software Inc., San Diego, CA). Linear regression analyses were conducted using Microsoft Excel 2000.
Results
Expression and Purification of HT-Cyp2d22 and HT-CYP2D6 Enzymes. High titer baculovirus stocks, T. ni suspension culture, and heme addition proceeded in a manner analogous to that used for CYP2C9 and CYP2D6 (Haining et al., 1996; Yu et al., 2001). Cultures exhibiting greater than 100 nM equivalents of active P450 enzyme were used for purification. Holoprotein yields were estimated by measuring carbon monoxide difference spectra. Both HT-Cyp2d22 and HT-CYP2D6 enzymes behaved in a chromatographically identical manner throughout the purification procedures. Contaminants were removed by passage through nickel resin. Highly purified, detergent-free P450 enzyme was collected after dialysis the fractions eluted from the column (Fig. 1). The total yields were 61.1% and 55.7% for HT-Cyp2d22 and HT-CYP2D6 enzymes, respectively. The carbon monoxide difference spectra for HT-Cyp2d22 isoform exhibited Soret maxima at 449 nm with no evidence of cytochrome P420 formation (Fig. 2). Meanwhile, all these samples were analyzed by Western blotting using an anti-CYP2D6 antibody. As expected, all the CYP2D protein products were detected in these samples, although the antibody is a polypeptide specific to CYP2D6 isoform (data not shown).

Typical CO difference spectrum of mouse HT-Cyp2d22 showing the maximal absorbance at 449 nm. Standard procedures (Omura and Sato, 1964) were followed.
ESI/LC-MS Analysis of HT-Cyp2d22 and HT-CYP2D6 Isoforms. We were able to determine the whole molecular mass of each purified P450 enzyme using ESI/LC-MS analysis to compare with their theoretical molecular masss. The experimental molecular masses were 60,287.5 Da and 59,430.0 Da for HT-Cyp2d22 and HT-CYP2D6 isoform, respectively, compared with the molecular mass values of 60,222.6 Da and 59,384.3 Da calculated according to their amino acid sequences. The differences between the experimental and predicted molecular masses were only 64.9 Da and 45.7 Da for HT-Cyp2d22 and HT-CYP2D6 proteins, suggesting that these proteins expressed in insect cells do not have any major post-translational modification, and such modification is unnecessary for their P450 activity.
Dextromethorphan O- and N-Demethylation. Dextromethorphan, an over-the-counter antitussive drug, is a widely used probe for polymorphic CYP2D6 activity both in vivo and in vitro, and possibly for CYP3A activity (Gorski et al., 1994; Ducharme et al., 1996; Jones et al., 1996; Krecic-Shepard et al., 1999; Yu and Haining, 2001). CYP2D6 catalyzes both dextromethorphan O- and N-demethylation, with a low and a high Km, respectively (Yu et al., 2001), suggesting CYP2D6 enzyme may also contain two substrate orientations or two active sites. Polyhistidine-tagged CYP2D6 also carried out these two reactions with two similar Km values, 4.2 μM and 4.8 mM, compared with 5.1 μM and 3.9 mM by nonpolyhistidine-tagged CYP2D6 (Table 1), indicating the addition of hexahistidine tag at the N terminus did not affect the drug-metabolizing activity of CYP2D6.
Estimated kinetic parameters for dextromethorphan O- and N-demethylation catalyzed by CYP2D6, HT-CYP2D6, HT-Cyp2d22, and CYP3A4 isozymes
Each data point used to calculate kinetic constants was based on an average of duplicate determinations. Enzyme Michaelis-Menten parameters and error estimates thereof were generated by nonlinear regression analysis (GraphPad Prism 3.02).
Dextromethorphan was then used to compare the difference in enzymatic activity between mouse Cyp2d22 and human CYP2D6 isoforms. To our surprise, dextromethorphan N-demethylation was the main pathway catalyzed by HT-Cyp2d22, whereas O-demethylation was the main metabolic pathway mediated by (non)HT-CYP2D6 at a comparably low substrate concentration (100–200 μM; Fig. 3). The calculated apparent constant value for dextromethorphan O-demethylation catalyzed by HT-Cyp2d22 was 250 μM, much higher than that by HT-CYP2D6 (Table 1). However, HT-Cyp2d22 catalyzed dextromethorphan N-demethylation with a Km value of 418 μM, much lower than the Km value of 4750 μM by HT-CYP2D6. Because dextromethorphan N-demethylation is known as CYP3A4-mediated biotransformation pathway, the catalytic activity of this reaction was compared between mouse Cyp2d22 and human CYP3A4. Interestingly, their apparent constant and intrinsic clearance values were very close, which were 418 μM and 48.7 nl/pmol P450/min for HT-Cyp2d22 and 302 μM and 81.1 nl/pmol P450/min for CYP3A4. We also detected the O-demethylated metabolites, dextrorphan, produced by Cyp2d22 (Fig. 3) and CYP3A4 isoforms and compared the kinetic parameters between them. Again, their apparent constant and intrinsic clearance values for dextromethorphan O-demethylation were very close: 171 μM and 32.5 nl/pmol P450/min for HT-Cyp2d22 and 157 μM and 34.3 nl/pmol P450/min for CYP3A4 (Table 1). Because this similarity in catalytic activity between mouse Cyp2d22 and human CYP3A4 isoforms was not expected, more substrates were selected to further compare the similarity in enzymatic activities between them.
Codeine O- and N-Demethylation. Codeine, whose backbone chemical structure is strikingly similar to that of dextromethorphan, is metabolized to morphine and norcodeine through O- and N-demethylation, respectively. In humans, codeine O- and N-demethylation are attributable to isoforms CYP2D6 and CYP3A, respectively (Caraco et al., 1996; Yue and Sawe, 1997). In our previous study on CYP2D6 pharmacogenetics, we found that all the CYP2D6 allelic variants catalyzed primarily codeine O-demethylation (Yu et al., 2002).
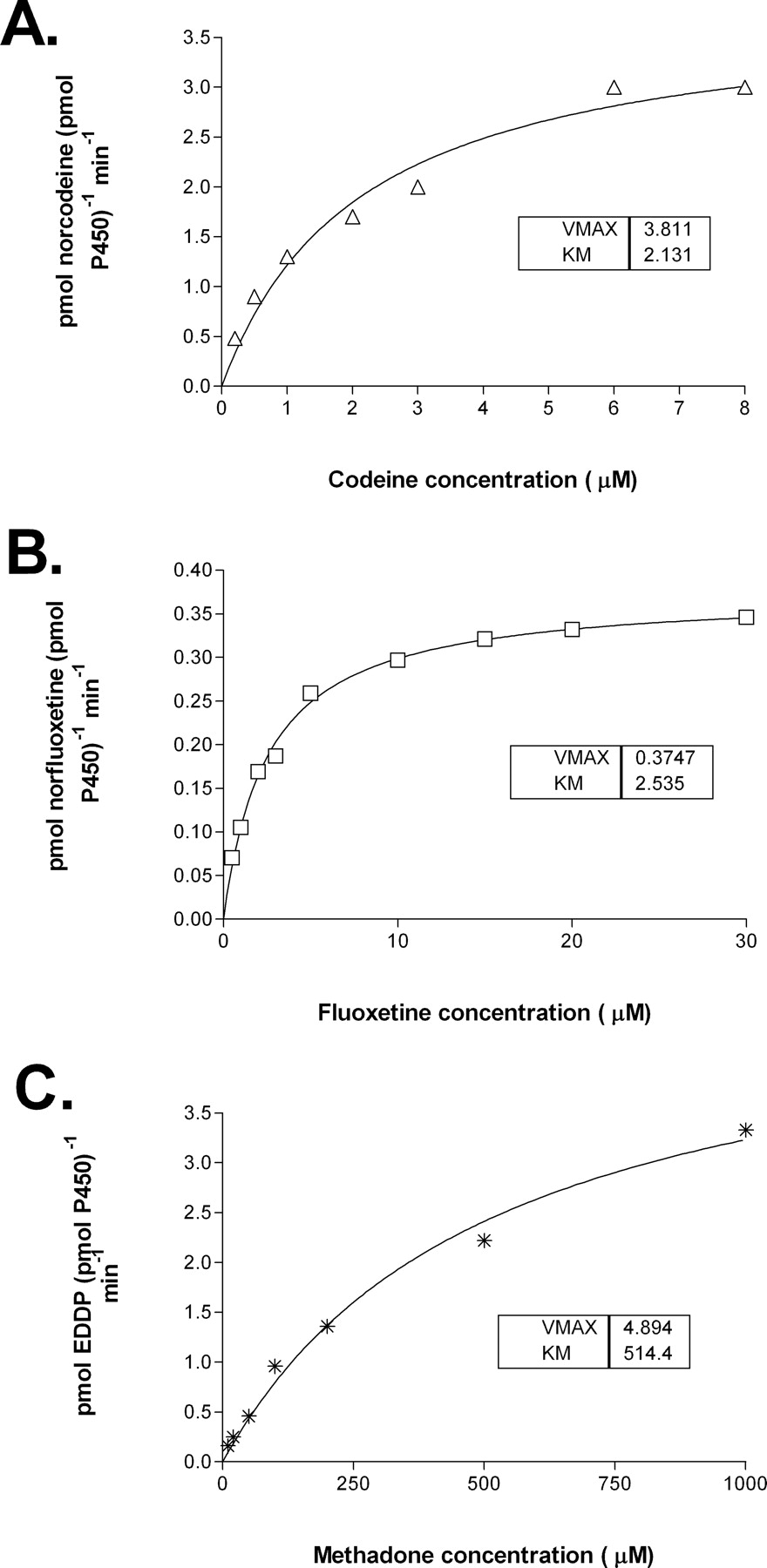
Morphine was produced by HT-CYP2D6 with a Km at 277 μM, which was close to that (178 μM) by unmodified CYP2D6 isoform. Norcodeine was not detected in the incubations with (non)HT-CYP2D6 enzyme. However, morphine was not detected in the incubation reactions containing HT-Cyp2d22 or CYP3A4, and norcodeine was produced as the only metabolite. The Vmax and Km values for codeine N-demethylation catalyzed by HT-Cyp2d22 were 3.8 pmol/pmol P450/min and 2150 μM, respectively. CYP3A4 proceeded codeine N-demethylation (Fig. 4) with a Km of 619 μM (Table 2), which was only about 3-fold lower than that by HT-Cyp2d22.
Calculated kinetic parameters for codeine, fluoxetine and methadone biotransformations catalyzed by CYP2D6, HT-CYP2D6, HT-Cyp2d22, and CYP3A4 isoforms
Each data point used to calculate kinetic constants was based on an average of duplicate determinations. Enzyme Michaelis-Menten parameters and error estimates thereof were generated by nonlinear regression analysis (GraphPad Prism 3.02).
Fluoxetine and Methadone N-Demethylation. Fluoxetine is a well known potent inhibitor of CYP2D6, as well as a substrate, which is N-demethylated to form norfluoxetine in humans. HT-Cyp2d22 catalyzed fluoxetine N-demethylation with an estimated Km value of 110 μM, much higher than the Km value of 2.5 μM and 6.6 μM mediated by HT-CYP2D6 and (non)HT-CYP2D6, respectively, but yet close to the Km value of 168 μM exhibited by CYP3A4 (Table 2; Fig. 4).
Methadone, also a potent inhibitor to CYP2D6, is N-demethylated by CYP3A4 to produce EDDP in humans (Iribarne et al., 1996; Moody et al., 1997; Oda and Kharasch, 2001). Unexpectedly, purified HT-Cyp2d22 also catalyzed this biotransformation reaction. The calculated apparent constant value of methadone N-demethylation by HT-Cyp2d22 was 517 μM, much higher than the Km value of 32.9 μM mediated by CYP3A4 (Table 2; Fig. 4).

Michaelis-Menten saturation curves for dextromethorphan O-demethylation (A) and N-demethylation (B) as catalyzed by recombinant Cyp2d22. Purified Cyp2d22 was reconstituted with lipid and rat P450-reductase as described under Materials and Methods before initiation.
Testosterone 6β-Hydroxylation. Steroid hydroxylation is a well established function of human CYP3A and murine Cyp2d isoforms. Testosterone 6β-hydroxylation, a widely used index reaction for CYP3A4 activity, was reported to be catalyzed by the partially purified mouse Cyp2d enzyme (Masubuchi et al., 1997) but not mediated by Cyp2d22-transfected cells (Blume et al., 2000). We believe this partially purified mouse Cyp2d enzyme was the Cyp2d22 isoform because its reported amino acid sequence at the N terminus was exactly the same as that of Cyp2d2 protein and different from that of any other known murine Cyp2d isoforms. To clarify this literature discrepancy and to further compare the substrate specificity between mouse Cyp2d22 and human CYP3A4, we examined whether the purified HT-Cyp2d22 could also catalyze testosterone 6β-hydroxylation reaction. As expected, 6β-hydroxytestosterone was detected in the incubation reactions with human CYP3A4 but not in the reactions with (non)HT-CYP2D6. Meanwhile, HT-Cyp2d22 did not mediate testosterone 6β-hydroxylation, which was consistent with the result obtained by Blume et al. (2000).

Michaelis-Menten saturation curves for codeine N-demethylation (A), fluoxetine N-demethylation (B), and methadone N-demethylation (C) as catalyzed by recombinant Cyp2d22.
Inhibition Analysis. CYP2D6 and CYP3A4 inhibitors were used to study and compare their inhibition potencies against the Cyp2d22 isoform. The mean Ki value of quinidine for dextromethorphan O-demethylation catalyzed by purified CYP2D6 enzyme was determined as 34 nM (Yu and Haining, 2001). Using HT-CYP2D6, the mean Ki value obtained for the dextromethorphan O-demethylation was 68 nM, indicating that the inhibition potency of quinidine to CYP2D6 enzymatic activity was not affected by the fusion of the hexahistidine tag. However, quinidine (5–500 nM) did not show strong inhibition (≤20%) to HT-Cyp2d22-catalyzed dextromethorphan O- or N-demethylation. Rather, a well known CYP3A4 inhibitor ketoconazole (100–300 nM) showed moderate inhibition (20–40%) against Cyp2d22-catalyzed dextromethorphan N- and O-demethylation (not shown).
Sequence Similarity of Cyp2d22 to Human CYP2D6 and CYP3A4. The predicted mouse Cyp2d22 protein consists of 505 amino acids and shares 74% amino acid sequence identity with human CYP2D6, a little greater than that with other known murine Cyp2d isoforms (69%, 69%, and 67% for Cyp2d9, Cyp2d10, and Cyp2d11, respectively; Fig. 5). Because the mouse Cyp2d22 enzyme showed certain enzymatic similarity as human CYP3A4, we then compared its sequence similarity with CYP3A4. As expected, the amino acid sequence of Cyp2d22 was less than 40% identical to CYP3A4. Nonetheless, the amino acid sequence of essential components, such as the cysteine-pocket, heme-binding domain with meander, and I, J helix were highly conserved among all the isoforms examined.
Discussion
At least 20 CYP2D genes have now been identified in mammals and humans (Kimura et al., 1989; Nelson et al., 1996; Mankowski et al., 1999; Blume et al., 2000; Hosseinpour and Wikvall, 2000). In humans, three P450 genes belonging to the CYP2D subfamily (CYP2D6, CYP2D7P, and CYP2D8P) have been identified. However, CYPD7P and CYP2D8P, which present typically in a given haplotype, are pseudogenes, and only CYP2D6 produces functional protein in human liver (Kimura et al., 1989). CYP2D1 through CYP2D5 and CYP2D18 are found in the rat, and Cyp2d9 through Cyp2d13 and Cyp2d22 are from the mouse (Nelson et al., 1996; Blume et al., 2000). The mouse Cyp2d22 gene contains 1515 bp and encodes a protein with 505 amino acids. Its DNA sequence shares 87 to 90% identity with rat CYP2D3 through CYP2D5 and CYP2D18, with a corresponding amino acid identity of 71 to 85%. Interestingly, the amino acid sequence of the predicted mouse Cyp2d22 is less similar to mouse Cyp2d9 (69%), Cyp2d10 (69%), or Cyp2d11 (67%) than human CYP2D6 (74%) and monkey CYP2D17 (75%; 43).
Most of these mammalian CYP2D enzymes are expressed in liver and are known to be responsible for the biotransformation of xenobiotics. Given the sequence similarity with CYP2D6, we reasoned that characterization of the enzymatic function for the encoded Cyp2d22 protein would contribute to the understanding of the CYP2D6-like phenotype or lack thereof in mice. Recently, Lofgren et al. (2004) reported CYP2D6-like enzymatic activity in mouse liver microsomes. This activity correlated with a protein that was immunoreactive to anti-rat Cyp2d4 antibody and that appeared more prevalent in females than males (Lofgren et al., 2004). However, Cyp2d22 is known to be highly expressed in both male and female mouse liver and is modestly expressed in a variety of other tissues, such as adrenal, ovary, and mammary glands (Blume et al., 2000). Thus, our results would suggest that Cyp2d22 is not the CYP2D isozyme responsible for the CYP2D6-like enzyme activity reported by Lofgren et al. (2004) in female mouse liver.

Alignment of the amino acid sequence of mouse CYP2D22 to human CYP2D6 and CYP3A4 proteins.
Mouse Cyp2d22, purified from cDNA-transfected insect cells and characterized by immunochemical and ESI/LC-MS analyses, indeed catalyzes the biotransformation of dextromethorphan and shows substantially decreased O-demethylase activity as compared with human CYP2D6 (Table 1). However, Cyp2d22 produces more N-demethylated metabolite, 3-methoxymorphinan, than O-demethylated metabolite, dextrorphan. The switch of the metabolic pathways of dextromethorphan metabolism suggests a difference of its enzymatic function from that of CYP2D6. Moreover, Cyp2d22 catalyzes dextromethorphan N-demethylation with more Km value than CYP2D6 but similar to that exhibited by CYP3A4 (Table 1). The functional similarity between Cyp2d22 and CYP3A4 is further confirmed by the results obtained from codeine, fluoxetine, and methadone biotransformation reactions (Table 2), although the identity of amino acid sequence is less than 40% between these isoforms. This similarity in substrate specificity between P450 isoforms across the families and species may provide some helpful information for the study of P450 orthogenesis. Nevertheless, it remains to be determined whether the enzymatic functions of mouse Cyp2d22 in vivo are largely similar to those of human CYP3A4.
Distinct enzymatic function between Cyp2d22 and CYP2D6 is presumably because of their own unique active sites distinct from each other. This is supported by our inhibition study, which shows that quinidine strongly inhibits CYP2D6-mediated drug metabolism, whereas it does not reduce Cyp2d22 enzymatic activity. In contrast, CYP3A4 selective inhibitor, ketoconazole, shows modest inhibition again Cyp2d22 activity. Knowledge of selective and potent inhibitors of the Cyp2d22 isoform could be of great benefit to future functional and molecular modeling studies involving the Cyp2d22 and other murine Cyp2d enzymes.
The presence of CYP2D isoforms in extrahepatic tissues, such as CYP2D6 and CYP2D18 in human and rat brains, respectively (Kawashima et al., 1996; Gilham et al., 1997; Voirol et al., 2000; Siegle et al., 2001), has been shown, where they may be involved in the biosynthesis and biodegradation of endogenous compounds. Indeed, CYP2D6 has been shown to mediate the catabolism of phenylalkylamines (Hiroi et al., 1998; Haining and Yu, 2003) and indolealkylamines (Yu et al., 2003). CYP2D18, which shares 85% amino acid sequence identity with Cyp2d22, has also been shown to catalyze dopamine oxidation (Thompson et al., 2000). CYP2D25, purified from pig liver, has shown vitamin D3 25-hydroxylase activity with an apparent Km of 0.1 μM, a concentration within the physiological range (Hosseinpour and Wikvall, 2000). No evidence indicates that Cyp2d22 metabolizes endogenous steroids of any kind (Blume et al., 2000). However, the partially purified mouse Cyp2d enzyme, which should be the Cyp2d22 protein according to the reported amino acid sequence, catalyzes a variety of testosterone hydroxylation reactions (Masubuchi et al., 1997). Our experiments reveal that neither Cyp2d22 nor CYP2D6 produces any hydroxylated metabolite from testosterone, whereas CYP3A4 does. This result is consistent with that reported by Blume et al. (2000) using Cyp2d22-transfected cells. In fact, no recombinant mouse or rat CYP2D isoforms have been found to catalyze the reaction to date. Nonetheless, further studies will be required to investigate whether murine Cyp2d isozyme catalyzes the biotransformation of endogenous compounds such as the neuroregulatory monoamines.
In summary, biochemical characterization, ESI/LC-MS analysis, and immunochemical confirmation of the mouse Cyp2d22 isozyme were achieved, following the purification from insect cells using a baculovirus-mediated expression system. Comparative kinetic analyses toward multiple substrate reactions and inhibition studies show that Cyp2d22 possesses deficient human CYP2D6-like activity but has a certain degree of similarity in enzymatic function as human CYP3A4. These results suggest that murine Cyp2d22 has substrate specificity intrinsically different from other mammalian CYP2D isozymes.
Footnotes
-
This work was supported in part by Grant ES09894 from the National Institute of Environmental Health Sciences.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008870.
-
ABBREVIATIONS: P450, cytochrome P450; ESI/LC-MS, electrospray ionization/liquid chromatography mass spectrometry; EDDP, 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine; PMSF, phenylmethylsulfonyl fluoride; HPLC, high-performance liquid chromatography; HT, hexahistidine-tagged.
- Received December 7, 2005.
- Accepted March 29, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}