


Abstract
Prasugrel, a prodrug, is a novel and potent inhibitor of platelet aggregation in vivo. The metabolism of prasugrel and the elimination and pharmacokinetics of its active metabolite, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid (R-138727), three inactive metabolites, and radioactivity were determined in five healthy male subjects after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel. Prasugrel was rapidly absorbed, and maximum plasma concentrations of radioactivity and R-138727 were achieved in 30 min, indicating rapid formation of R-138727. Prasugrel was extensively metabolized in humans, first by hydrolysis to a thiolactone, followed by ring opening to form R-138727, which was further metabolized by S-methylation and conjugation with cysteine. Total radioactivity was higher in plasma than in blood, suggesting limited penetration of prasugrel metabolites into red blood cells. Approximately 70% of the dose was excreted in the urine and 25% in the feces.
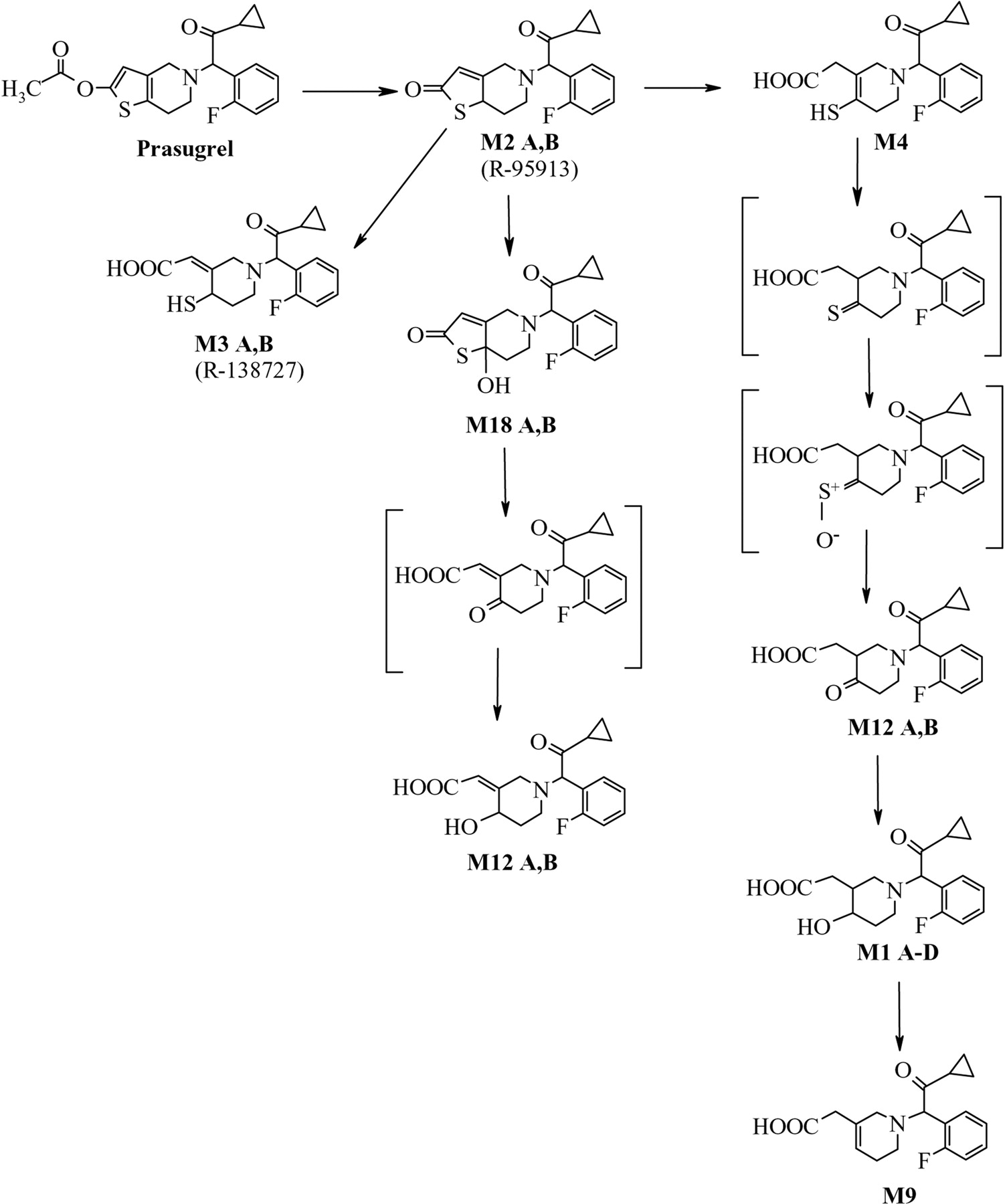
Clopidogrel and ticlopidine are thienopyridine prodrugs that are activated in vivo to pharmacologically active metabolites that bind irreversibly to the platelets' P2Y12 receptor, thus inhibiting platelet aggregation. Studies with [14C]clopidogrel showed that the major metabolic pathway in humans is hydrolysis of clopidogrel to an inactive acid analog; no further metabolites were reported (Lins et al., 1999). Prasugrel (Fig. 1) is a novel and potent thienopyridine prodrug that also inhibits platelet aggregation in vivo. The structure of the thiol-containing active metabolite of prasugrel, R-138727, was previously reported (Sugidachi et al., 2000, 2001). Similarly, the structure of the thiol-containing active metabolite of clopidogrel was determined by in vitro studies (Pereillo et al., 2002). The platelet-inhibitory activity of prasugrel and initial pharmacokinetic data were recently reported (Jakubowski et al., 2006). In vivo, prasugrel is rapidly hydrolyzed to a pharmacologically inactive thiolactone (R-95913), followed by cytochrome P450-dependent ring opening to form the active metabolite R-138727 (Rehmel et al., 2006). The active metabolite of prasugrel possesses two chiral centers, and its four isomers were shown to possess varying degrees of activity toward inhibition of platelet aggregation (Hasegawa et al., 2005).
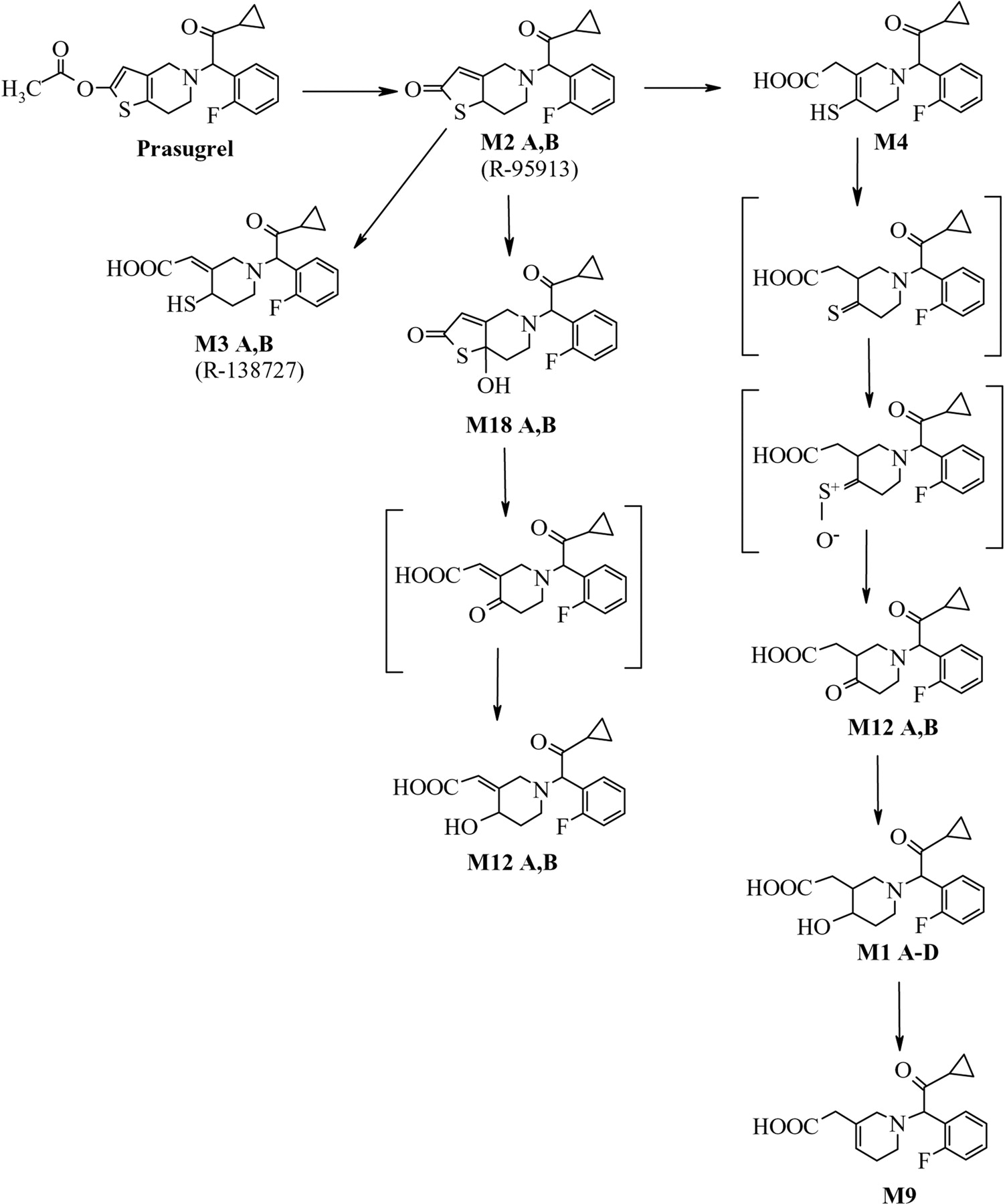
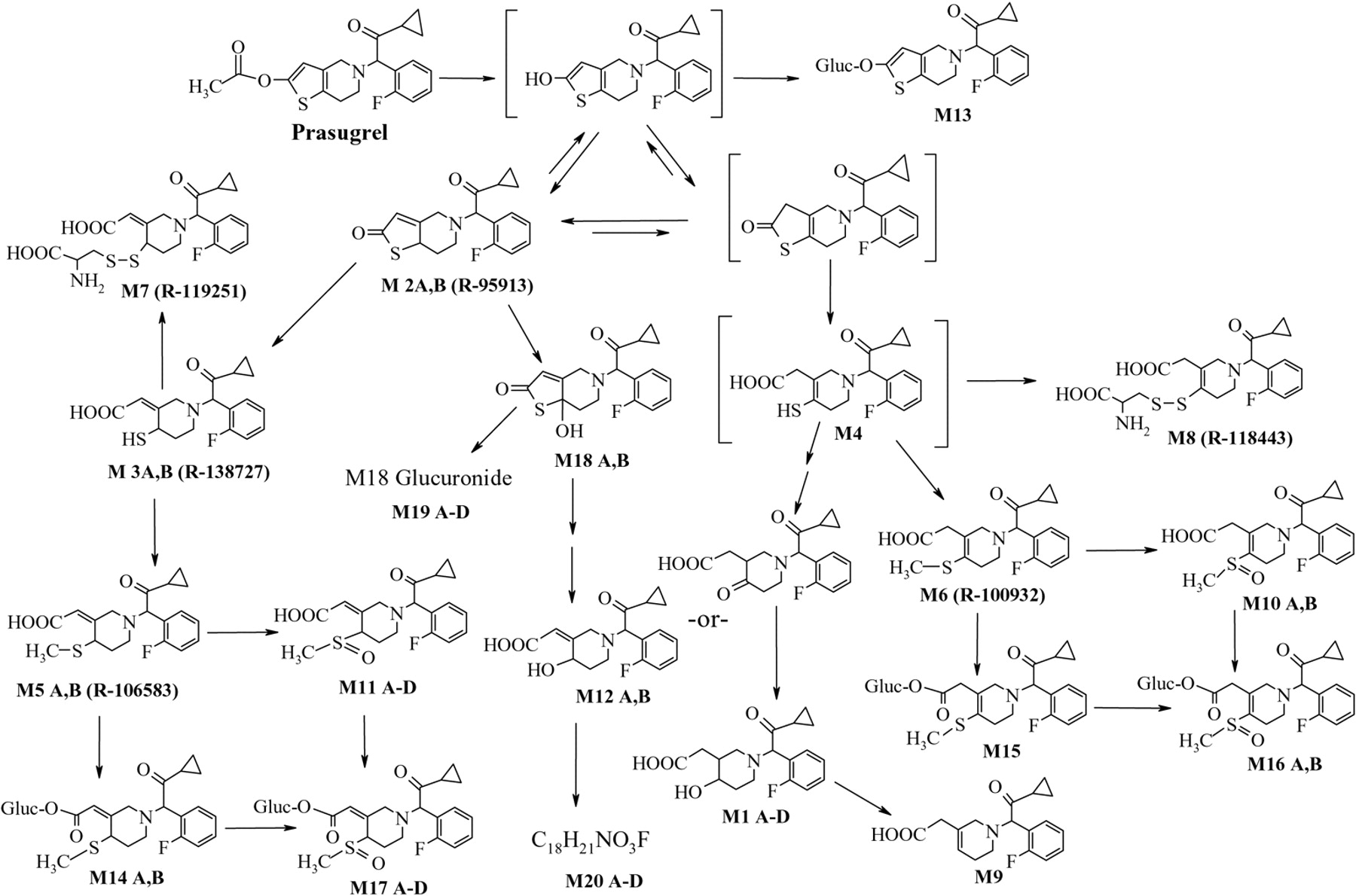
The prasugrel metabolites measured in human plasma in initial studies (Asai et al., 2006) indicated that after formation of R-95913, two thiol-containing compounds are formed, R-138727 and M4, which are further metabolized by S-methylation to R-106583 and R-100932, or conjugation with cysteine to R-119251 and R-118443 (Fig. 1). Compounds R-95913, R-106583, and R-100932 were measured in plasma as indicators for absorption and exposure to prasugrel and its active metabolite. In this report, the physiologic disposition of prasugrel in healthy subjects following a 15-mg (100 μCi) p.o. dose of [14C]prasugrel is presented.
Materials and Methods
Radiolabeled Drug and Chemicals. Prasugrel hydrochloride was provided by Sankyo Co., Ltd. (Tokyo, Japan). [14C]prasugrel (radiochemical purity 99.3%) was synthesized at Amersham Biosciences (Whitchurch, Cardiff, UK). Standards of the metabolites M1-A, M1-B (UBS-1767), M1-C, M1-D (UBS-1766), M2 (R-95913), M3 (R-138727), M5 (R-106583), M6 (R-100932), M7 (R-119251), and M8 (R-118443) were provided by Sankyo Co., Ltd. β-Glucuronidase (Helix pomatia) was purchased from Sigma-Aldrich (St. Louis, MO). Permafluor V and Aquassure were purchased from PerkinElmer Life Sciences (Boston, MA). Beckman Protein Plus+ was purchased from Beckman Coulter Inc. (Fullerton, CA). All the other reagents used in these experiments were either of analytical or high-performance liquid chromatography (HPLC) grade.
Human Study. The study was approved by the appropriate ethical review boards and conducted in accord with the Declaration of Helsinki and is consistent with applicable Good Clinical Practice guidelines. All the subjects provided written informed consent. This was a single-center, open-label, single-dose study. The subjects were judged to be in good health by screening evaluation, which included their medical history, a complete physical examination, and clinical laboratory tests. Five male subjects, three Caucasians and two of African descent, between the ages of 31 to 60 years (average, 43 years), and weighing 65.6 to 92.1 kg (average, 76.1 kg) participated in the study.

Proposed metabolic scheme of prasugrel in humans.
The [14C]prasugrel dose, administered after an overnight fast, consisted of prasugrel hydrochloride and 14C-labeled prasugrel base dissolved in 170 ml of degassed cola with 2% ethanol, to provide 15 mg of prasugrel with approximately 100 μCi of radioactivity. The dose container was rinsed with an additional 80 ml of the vehicle solution that was also swallowed. The actual dose administered was determined to be 14.7 mg of prasugrel containing 90 ± 3.6 μCi. Only water and decaffeinated coffee were allowed for the first 5 h after dosing. The subjects remained in the clinical facility for 2 weeks after the dose and were monitored throughout that time.

Mean (±S.D.) plasma concentrations of radioactivity (•), R-106583 (○), R-138727 (□), R-119251 (▴), and R-95913 (⋄) after a single 15-mg p.o. dose of [14C]prasugrel.
Collection of Biological Samples. Serial venous blood samples were collected for pharmacokinetic analysis in EDTA-containing tubes before drug administration and starting 15 min and ending 96 h after the dose for the assay of radioactivity in blood and plasma and for the determination of the plasma concentrations of prasugrel inactive metabolites R-95913, R-106583, and R-119251. For the analysis of the active metabolite R-138727, separate blood collections (3 ml each) were made in EDTA tubes at the same times as those for the inactive metabolites; 25 μl of a 0.5 M 3′-methoxyphenacyl bromide solution in acetonitrile was added within 30 s, and the contents were mixed. Addition of the derivatizing reagent to the blood sample rapidly after collection and before plasma separation was required for accurate determination of the active metabolite concentration (Farid et al., 2007). Plasma, separated by centrifugation within 30 min, was frozen at –70°C until analysis for the active and inactive metabolites of prasugrel. Additional blood samples were collected (with and without derivatization) between 0.5 and 12 h after the [14C]prasugrel dose for metabolite profiling.
Urine and feces were collected before dosing (control samples) and at predetermined intervals until an insignificant amount of radioactivity (≤0.3% of the 14C dose) was excreted in a 24-h collection interval. Breath samples were collected from each subject at 1 and 1.5 h postdose and counted for determination of expired 14CO2.
Determination of Prasugrel Metabolites in Plasma. Plasma concentrations of R-138727, R-95913, R-106583, and R-119251 were determined by validated liquid chromatography/tandem mass spectrometry (LC/MS/MS) methods described previously (Farid et al., 2007).
Determination of Radioactivity. Plasma, urine, feces, and breath concentrations of radioactivity were determined by liquid scintillation counting (LSC). Plasma and urine samples (1 ml each) were counted for 14C content after the addition of the liquid scintillation mixture. Triplicate feces homogenate and blood samples (approximately 0.5 g each) were placed into combustion thimbles and weighed. The samples were allowed to air dry overnight and then were combusted in a Packard Tricarb Oxidizer 307 (PerkinElmer Life Sciences). The resulting 14CO2 was trapped and assayed for radioactivity using LSC. All the counting data were automatically corrected for counting efficiency using external standardization method.

Mean (± S.E.M.) of the percentages of 14C dose excreted in humans following a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel (•, feces; □, urine; ▴, 14C).

Radiochromatographic profile of 0.5-h human plasma after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel before (top) and after (bottom) derivatization of blood with 3-methoxyphenacyl bromide.
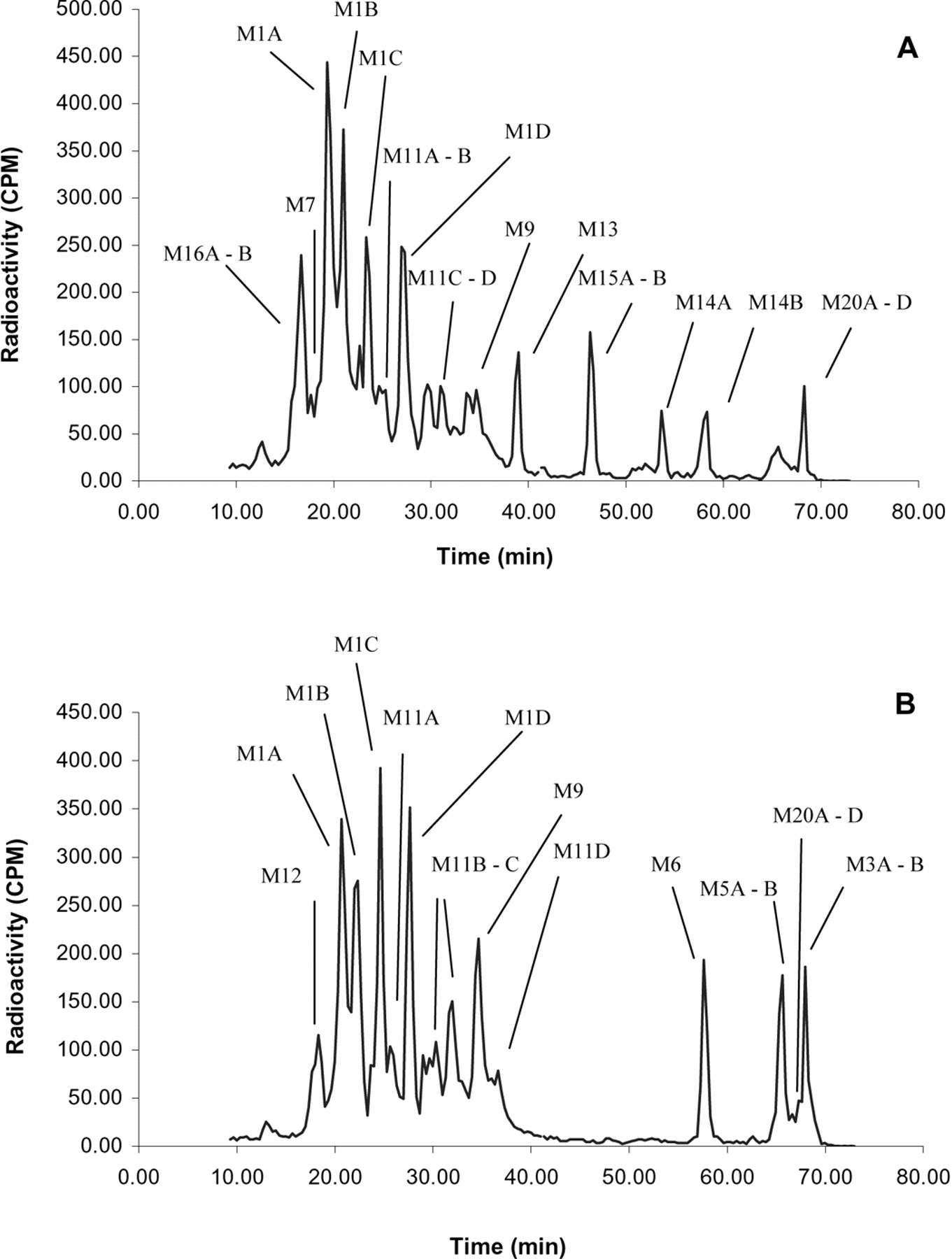
A, radioprofile of pooled 0- to 24-h human urine (unhydrolyzed). B, radiochromatographic profile of pooled 0- to 24-h human urine after hydrolysis with β-glucuronidase.

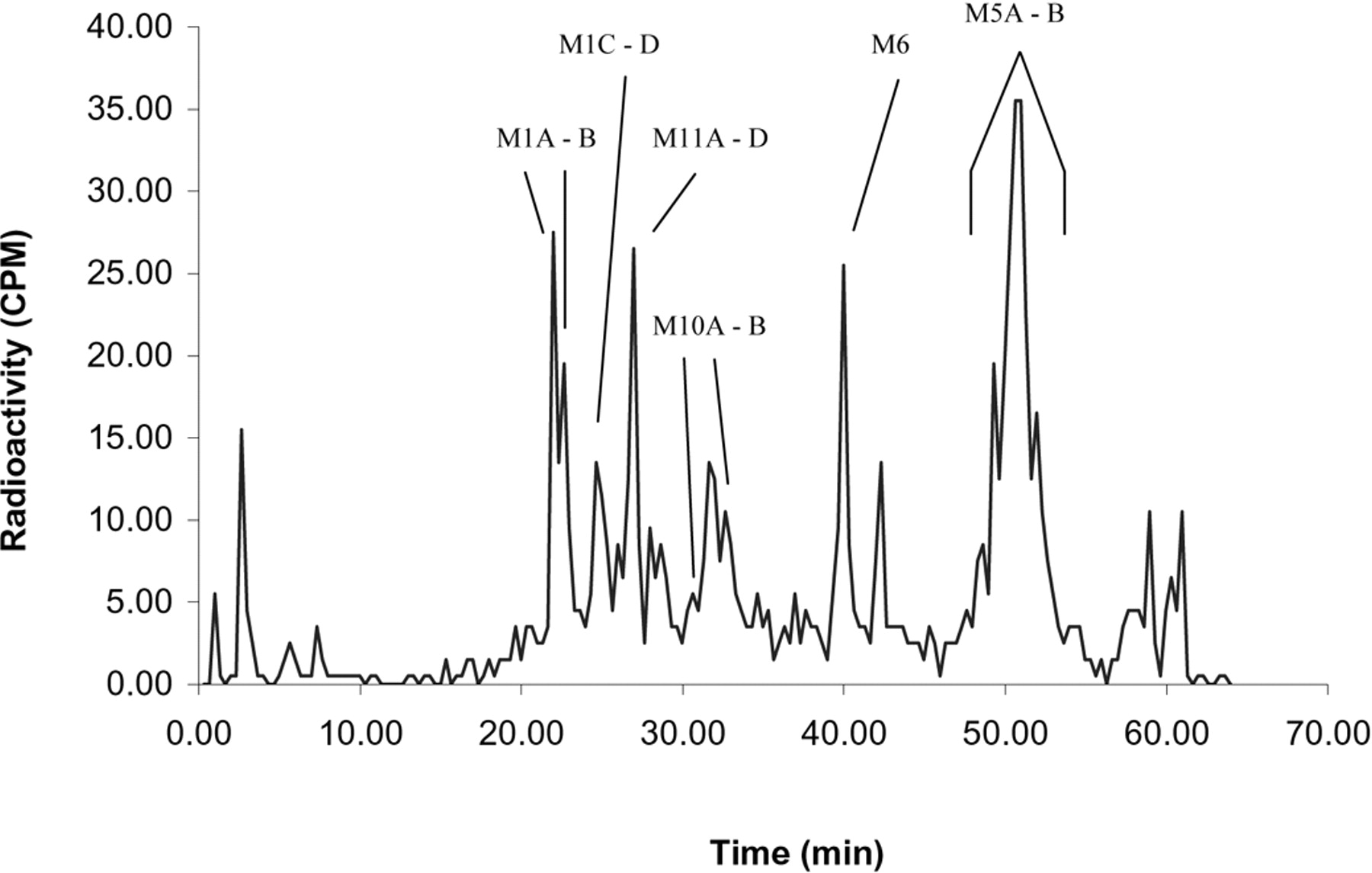
Radiochromatographic profile of a 24-h human fecal extract after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel.
The minimum detectable activity for plasma, blood, urine, and feces samples ranged from 0.37 to 0.52 ng-Eq/assayed matrix.
Pharmacokinetic Analysis. Each subject's plasma concentrations of radioactivity and the four prasugrel metabolites versus time were analyzed by a noncompartmental method. The observed peak plasma concentrations (Cmax) and time to reach Cmax (Tmax) were reported. The area under the plasma concentration versus time curve (AUC) was calculated from time 0 to the time of the last quantifiable plasma concentration. The pharmacokinetic parameters for radioactivity in blood were similarly determined.
Sample Preparation for Chromatographic Analysis. Plasma samples (0.5 ml) were extracted consecutively with 1 ml of 0.2% formic acid in CH3CN, 1 ml of methanol, and finally with 1 ml of 0.2% formic acid in CH3CN. The combined supernatants (3 ml) were evaporated to dryness under N2 and reconstituted in 0.3 ml of 0.2% HCOOH/CH3OH/CH3CN (7:1.5:1.5). The mean (±S.D.) extraction efficiency of radioactivity from plasma collected at 0.5, 1, 2, and 4 h after the dose was 96 ± 7%. The mean extraction efficiency of the 12-h plasma samples was 70 ± 1%.
Urine samples were directly chromatographed. Several urine samples were also hydrolyzed with β-glucuronidase, and 1-ml aliquots were subjected to solid-phase extraction using 1-cc Oasis HLB cartridges (Waters, Milford, MA). The cartridges were washed with 1 ml of water and 1 ml of 5% methanol in water. The radioactivity in the hydrolyzed urine was eluted with 1 ml of methanol. The methanol extracts were dried under N2, and the residues were reconstituted in 0.3 ml of water/CH3OH/CH3CN (7:1.5:1.5 v/v) for injection onto the HPLC system. The mean (±S.D.) extraction efficiency of radioactivity from hydrolyzed urine was 88 ± 3%.
Aliquots of fecal samples homogenates, ∼0.5 to 0.7 g, were extracted three times with 0.2% formic acid in CH3CN (3 ml each). The combined extracts of each feces sample was evaporated under N2 and reconstituted in 0.5 ml of 0.2% CH3COOH/CH3OH/CH3CN (7:1.5:1.5). The mean (±S.D.) extraction efficiency of radioactivity from feces was 61 ± 10%.
LC/Mass Spectrometry and HPLC Chromatographic Conditions for Radioactivity Profiling. A Shimadzu HPLC system (Shimadzu Corp., Kyoto, Japan) consisted of two model LC-10AD pumps, a SIL-10A autosampler, a DGU-3A degasser, and a model SCL-10A controller. HPLC radioprofiles of pooled plasma, pooled urine, and feces samples were obtained using microplate solid scintillation counting. Plasma and feces samples were analyzed using a Supelco Discovery C18 column (5 μm, 4.6 mm × 15 cm) (Sulpelco, Bellefonte, PA) and a gradient of 0.2% formic acid in water and CH3CN. After the initial 3 min, the CH3CN concentration in the mobile phase increased from 5% to 20% over 37 min. Five minutes later, the CH3CN concentration was again increased to 60% over 5 min and then to 90% 4 min later. The CH3CN concentration was held at 90% for 4 min before ending the 59-min run. The flow rate was 0.7 ml/min, and the column temperature was ambient. Urine samples were analyzed using a Supelco Discovery HSF5 column (5 μm, 4.6 mm × 25 cm) (Phenomenex, Torrance, CA) and a gradient of 10 mM ammonium acetate and CH3CN with a flow rate of 1 ml/min at ambient temperature. The CH3CN concentration in the mobile phase increased from 5% to 10% over the initial 3 min to 20% over the next 57 min, and then to 90% over the subsequent 8 min. These conditions were held for 4 min, ending the run at 72 min. The analytical column effluent was collected at 20-s intervals into a 96-well solid scintillant-coated plates (96-deep well LumaPlate, Perkin Elmer Life Science) for up to 64 (plasma and feces) or 74 min (urine). The plates were dried under centrifugal vacuum and counted on a Packard Top-Count-NXT counter (PerkinElmer Life Sciences) for 8 min/well. The averaged background was subtracted from the measured radioactivity for each well, and the results were plotted (counts versus time) to obtain a profile of the radiolabeled prasugrel metabolites. Peak areas were determined for the components in the chromatogram from the resulting counts.
LC/Mass Spectrometry and LC/MS/MS Analysis. The prepared plasma, feces, and urine samples were analyzed for prasugrel and its metabolites by LC/mass spectrometry (MS) and LC/MS/MS using a Finnigan LCQ DECA (Thermo Electron Corp, Somerset, NJ) mass spectrometer in positive ion electrospray mode. The HPLC column effluent was coupled to the mass spectrometer via a splitting tee. The chromatographic conditions were the same as those described above for HPLC profiling. The capillary heater was set to 225°C, and the spray voltage was 5 kV. For MS/MS experiments, the relative collision energy was set at 25%.
Hydrolysis with Dithiothreitol. To determine whether the unextractable plasma radioactivity was the result of disulfide bond formation between prasugrel active metabolite and plasma proteins, plasma samples were treated with dithiothreitol (DTT). Aliquots (200–250 μl) of the 12-h plasma from four of the five subjects (because of insufficient sample volume for remaining subject) were placed into two separate plastic vials. To one set of samples, phosphate buffer (0.3 M, pH 7.4, 20 μl) and 150 μl of 0.1 M DTT solution were added to each plasma aliquot (DTT-treated set). The samples were mixed, and an additional 50 μl of 0.1 M DTT solution was added 30 min later. The reacted plasma was allowed to remain standing for an additional 5 min. To the second set of samples (control set), 20 μl of the phosphate buffer and 200 μl of water were added. The samples were mixed and allowed to stand for 35 min. All the samples were extracted with 1 ml of 0.2% formic acid in CH3CN, vortexed, and centrifuged in a microcentrifuge at 13,000 rpm for 2 min. The extraction procedure was repeated with an additional 0.5 ml of 0.2% formic acid in CH3CN. The percentage of radioactivity recovered was determined in the supernatants from both control and DTT-treated extracted samples by LSC. The sample quantity and level of measurable radioactivity were too low to allow further HPLC radioprofiling and metabolite identification.

Mass spectrum of metabolite M2 after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel.
Results
A single 15-mg p.o. dose of prasugrel to five healthy male subjects was well tolerated, and no adverse reactions were reported in this study. Prasugrel was rapidly absorbed and metabolized in humans and was not detected in plasma. Tmax of radioactivity and most of the key metabolites was 0.5 h (Fig. 2; Table 1). Table 1 also provides the derived pharmacokinetic parameter estimates of radioactivity and the four prasugrel metabolites measured. On a molar basis, the four key metabolites, R-95913, R-138727, R-119251, and R-106583, comprised 40 ± 7% of the radioactivity AUC0–12 and 61 ± 21% of the radioactivity concentration in the plasma at 0.5 h after the dose. The median elimination half-life values for the measured metabolites ranged from 3 to 9 h, whereas that of plasma radioactivity was 188 h. The AUC of radioactivity in blood was approximately 63 ± 11% of that in plasma, indicating limited penetration of prasugrel's metabolites into red blood cells. No radioactivity was detected in the breath samples.
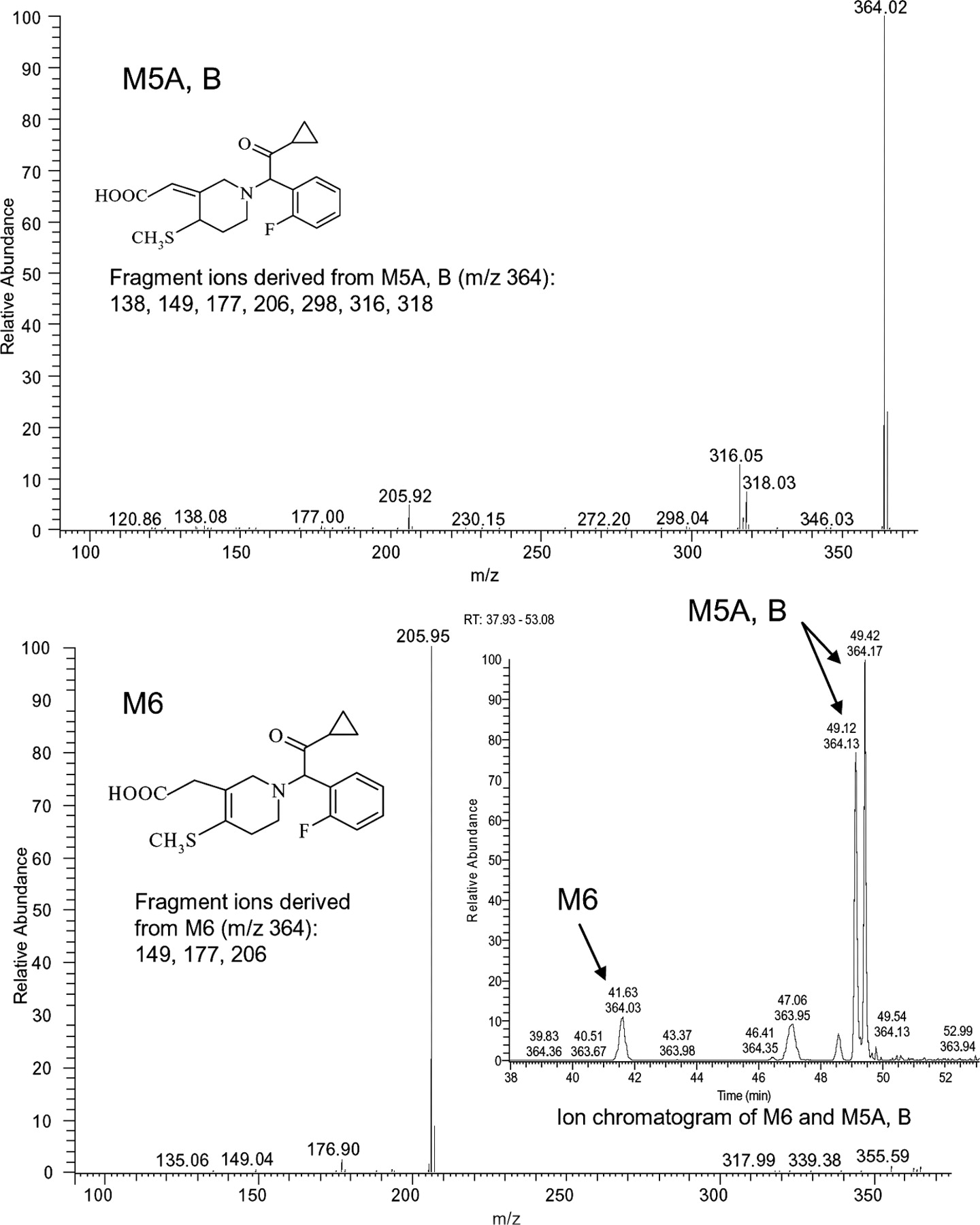
Structures and MS/MS spectra of metabolites M5-A, M5-B, and M6 from 2-h human plasma. The metabolites' ion chromatogram is shown in inset.
Mean pharmacokinetic parameters of prasugrel metabolites in humans after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel
Within 24 h, 61 ± 8% (mean ± S.D.) of the radioactivity from the [14C]prasugrel dose was excreted in urine, increasing to 68 ± 7% in 10 days. Excretion in the feces accounted for 27 ± 2% of the dose. The total recovery was 95 ± 7% of the administered 14C dose (Fig. 3). Neither prasugrel nor its esterase-hydrolyzed product M2 (R-95913) was detected in urine. Prasugrel and R-95913 (M2), which is mainly produced in the intestine, were not detected in feces; only metabolites of these two compounds were found in the feces. This suggests that the prasugrel dose was essentially fully absorbed and metabolized before excretion.
Prasugrel metabolites in plasma, urine, and feces were identified by radiochromatographic profiling and mass spectral analysis (Figs. 4, 5, 6). Figure 4 also shows the radiochromatograhic profile of plasma obtained after derivatization of the blood with 3′-methoxyphenacyl bromide to allow the detection of thiol-containing metabolites. The metabolite M4 was not detected in plasma after the 15-mg p.o. dose of prasugrel; however, its downstream metabolites M6 and M8 were observed (Fig. 1). Hydrolysis of the urine with β-glucuronidase resulted in the disappearance of several peaks from the radiochromatographic profile, confirming that the peaks that disappeared were glucuronic acid conjugates of prasugrel metabolites (Fig. 5).
Table 2 shows the characteristic productions (m/z) for prasugrel metabolites detected in the various matrices. Figures 7 and 8 show the structure and MS/MS spectra of the metabolites M2 (R-95913), M5 (R-106583), and M6 (R-100932) found in human plasma. Figures 9 and 10 show the structures and ion chromatograms of the standard isomers of M1 and as determined in the urine, respectively. The chromatographic profile and quantitation analysis showed that exposure was highest for the isomers of M5 (R-106583) in human plasma. The isomers of M1 were the major urinary metabolites and collectively accounted for 35% of the radioactivity in the urine and 21% of the prasugrel dose. The six metabolites identified in feces were also observed in plasma. The major metabolites were the isomers of M5 (R-106583) and M1, representing the majority of radioactivity excreted in the feces (Table 5; Fig. 6). Tables 3, 4, and 5 provide the mean percentages of the metabolites observed in plasma, urine, and feces, respectively.
Metabolites of prasugrel identified by LC/MS in human plasma, urine, and feces after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel

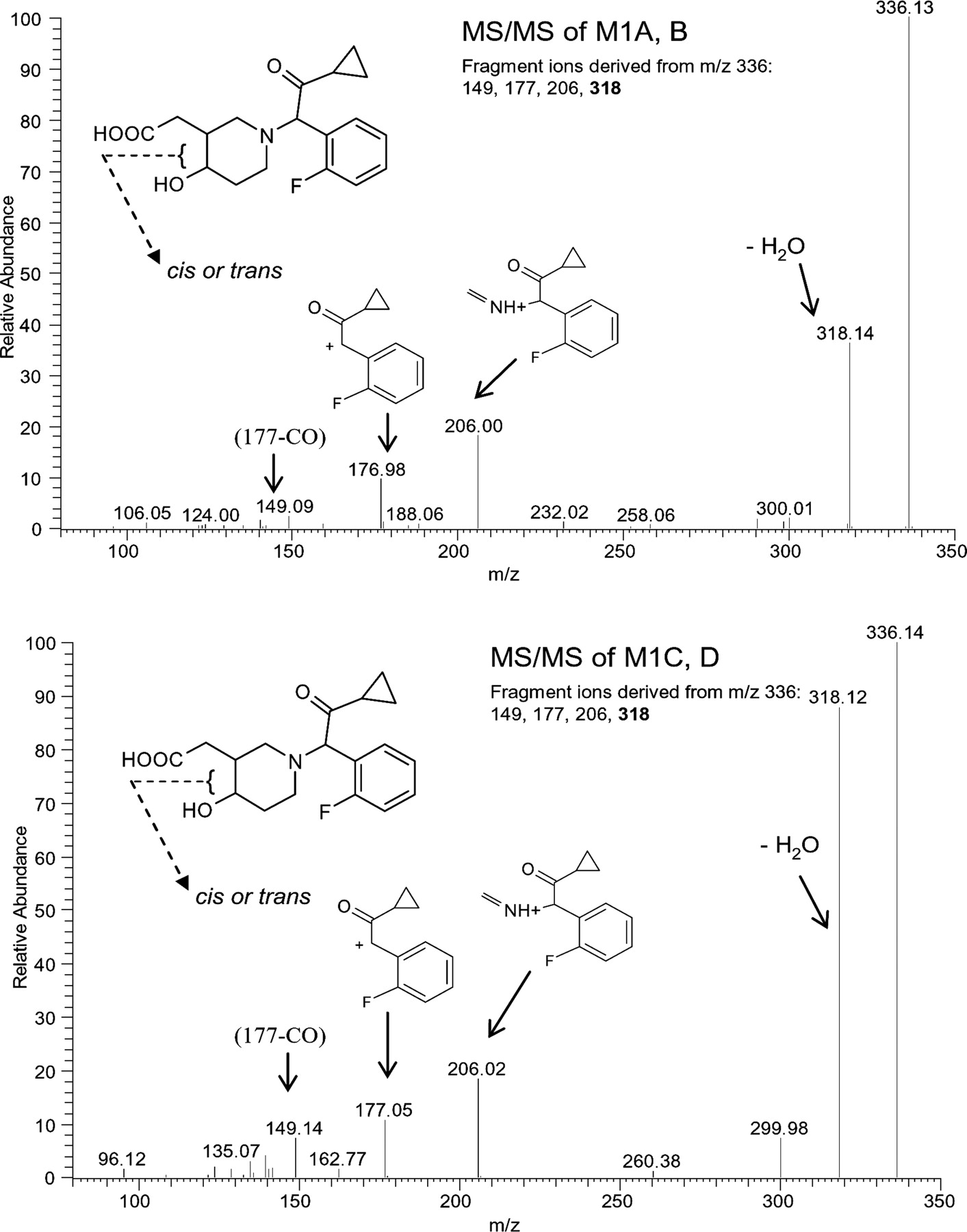
Structure and MS/MS spectra of metabolite M1 with synthetic standards for M1-A, -B (A) and M1-C, -D (B).
Mean percentages of metabolites (percentage of dose) present in 0- to 72-h human feces after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel
Mean (± S.D.) plasma concentration of prasugrel metabolites in human plasma after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel
Mean percentages of metabolites (percentage of dose) present in 0- to 24-h human urine after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel
Discussion
This is the first report describing the disposition of a thienopyridine in humans. Prasugrel is rapidly absorbed and extensively metabolized in humans; it is not detected in any of the matrices analyzed. The first step in prasugrel metabolism is the formation of the thiolactone, R-95913, which is formed by hydrolysis of prasugrel by intestinal and plasma esterases. Presence of prasugrel or R-95913 in the feces would have suggested that a portion of the prasugrel dose was not absorbed. However, because neither prasugrel nor R-95913 was detected in human feces, and only metabolites formed oxidatively (M1) and by S-methylation of the active metabolite (M5 from M3), it was concluded that the absorption of prasugrel after a p.o. dose is essentially complete.
The appearance of R-138727 (M3) in plasma within 15 min of dosing and achieving maximum plasma concentration by 30 min after the dose strongly suggest an important role for intestinal CYP3A in its formation from R-95913 and explain the rapid pharmacodynamic response observed after a prasugrel p.o. dose (Jernberg et al., 2006; Rehmel et al., 2006).
R-106583 is the major prasugrel metabolite found in human plasma, followed by R-138727 and R-95913 (Table 3). R-106583 is the only metabolite that represented >10% of the plasma radioactivity (Tables 1 and 3). The contribution of other metabolites was smaller. At 24 h postdose, the only quantifiable metabolite was R-106583, with a mean concentration of 7.9 ng/ml, whereas the mean plasma radioactivity was 46.8 ng-Eq/ml. The terminal half-life of plasma radioactivity (median, 188 h; range, 68.9–228 h) was longer than the terminal half-life of R-106583 (median, 8.7 h; range, 6.6–10.7 h). Although the half-life of plasma radioactivity should be interpreted cautiously, these data suggest that unmeasured and/or covalently protein-bound metabolites persist in the circulation. Hydrolysis of aliquots of the 12-h plasma samples with DTT released some of the protein-bound radioactivity (approximately 10–17%); however, the levels were too low to quantitate reliably.

A, ion chromatogram of metabolite M1 after a single 15-mg (100 μCi) p.o. dose of [14C]prasugrel. B, ion chromatogram of synthesized metabolite M1-C, -D (Standard UBS 1766). C, ion chromatogram of synthesized metabolite M1-A, -B (Standard UBS 1767).
As mentioned earlier, of the sulfhydryl compounds formed by opening the thiolactone ring of M2, only the enantiomers of R-138727 are pharmacologically active. The relative proportion of downstream metabolites of the thiol-containing compounds (R-106583 and R-119251 to R-100932 and R-118443) clearly indicates that the pathway leading to the formation of the active metabolite, R-138727, predominates in humans.
Accurate mass of the metabolites shown in Fig. 1 and Table 2 and the fragmentation information provided the basis for the proposing the structures shown. In particular, m/z 206 and its product m/z 177 (see Fig. 9) were present in all the metabolites, indicating that changes in the structure caused by metabolism of prasugrel did not involve that portion of the molecule. Enzymatic hydrolysis provided additional information regarding the glucuronide conjugates (M13, M19, M14, M17, M15, and M16). Unfortunately, the concentrations of the majority of prasugrel metabolites were too low to permit further isolation and/or analysis for absolute structural confirmation (e.g., by NMR).
Renal excretion was the major route for elimination of prasugrel metabolites in humans. The major metabolites observed in the urine were the diastereomers of M1 (m/z 336). The isolation of the four diastereomeric peaks is further confirmation of the postulated structure for this metabolite. As in prasugrel, the chiral carbon next to the nitrogen atom racemizes rapidly in vivo. However, the chirality of the hydroxyl group of M1 is preserved. Radiochromatographic profiling and MS showed that metabolites M1-A and M1-B were interconvertible and metabolites M1-C and M1-D were interconvertible (i.e., in each pair the two enantiomers contain the same hydroxyl group configuration).
The major metabolite detected in urine (and a minor one in plasma), M1, is a hydroxyl compound that does not contain a sulfur atom. Its formation suggests that a thione (derived from M4) is metabolized to a ketone (M12 ketone), which is then reduced to form M1. A possible pathway for the formation of M1 is shown in Fig. 11. The pathway for the cytochrome P450-catalyzed oxidation of thiones to form ketones, possibly through the formation of a sulfine (S+-O–), is not common but has been described previously (Madan and Faiman, 1994; Ortiz de Montellano, 1999). The enzymes responsible for this pathway have not been identified. Glutathione and glutathione S-transferase were shown to be involved in this metabolic pathway for some thiones (Madan et al., 1994). It is proposed that an M12 alcohol is formed as a result of hydrolysis of M18 and reduction of the formed ketone.
Proposed pathways for the formation of prasugrel metabolites M1, M9, and M12. (Structures in brackets are proposed intermediates that were not detected in urine, plasma, or feces samples.)
Conclusions
Prasugrel is essentially completely and rapidly absorbed and extensively metabolized in humans. Urine is the major pathway for the excretion of prasugrel metabolites, accounting for approximately 70% of the dose. The recovery of radioactivity from a single 15-mg [14C]prasugrel p.o. dose was 95%. The active metabolite of prasugrel (R-138727) was rapidly formed with a median Tmax of 0.5 h. The plasma 14C-terminal half-life (approximately 8 days) was longer than that of any of the metabolites measured in this study.
Prasugrel is rapidly hydrolyzed, forming the thiolactone M2, and is not detected in any of the matrices analyzed. The major metabolic pathway following hydrolysis of prasugrel in humans is the ring opening of the thiolactone to form the sulfhydryl compound M3 (R-138727), which undergoes further methylation and/or conjugation with cysteine. The other metabolites formed are essentially oxidative and conjugation products of the thiolactone or thiol-containing compounds.
Acknowledgments
We thank Julie Sherman of Eli Lilly and Company for the figure creation and administrative assistance in the preparation of this manuscript.
Footnotes
-
A portion of this work was presented at the 13th North American ISSX/20th JSSX Meeting, Maui, Hawaii, 2005 and was published in an abstract form in Drug Metab Rev37 (Suppl 2):86.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.014522.
-
ABBREVIATIONS: prasugrel, (±)-2-[2-acetyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone; R-138727, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid; R-95913, 2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone; R-106583, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-(methylthio)-3-piperidylidene]acetic acid; R-119251, (Z)-4-[(R)-2-amino-2-carboxyethyldisulfanyl]-3-carboxymethylidene-1-(α-cyclopropylcarbonyl-2-fluorobenzyl)piperidene; R-118443, 4-[(R)-2-amino-2-carboxyethyldisulfanyl]-3-carboxymethyl-1-(α-cyclopropylcarbonyl-2-fluorobenzyl)-1,2,5,6-tetrahydropyridine; R-100932, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-(methylthio)-1,2,5,6-tetrahydropyridin-3-yl]acetic acid; HPLC, high-performance liquid chromatography; LC/MS/MS, liquid chromatography/tandem mass spectrometry; LSC, liquid scintillation counting; AUC, area under the plasma concentration versus time curve; MS, mass spectrometry; DTT, dithiothreitol.
- Received January 5, 2007.
- Accepted March 29, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}