


Abstract
Covalent binding of reactive electrophilic intermediates to proteins is considered to play an important role in the processes leading to adverse drug reactions and idiosyncratic drug reactions. Consequently, both for the discovery and the development of new drugs, there is a great interest in sensitive methodologies that enable the detection of covalent binding of drugs and drug candidates in vivo. In this work, we present a strategy for the generation and analysis of drug adducts to human serum albumin. Our methodology is based on the isolation of albumin from blood, its digestion to peptides by pronase E, and the sensitive detection of adducts to the characteristic cysteine-proline-phenylalanine (CPF) tripeptide by liquid chromatography/tandem mass spectrometry. We chose acetaminophen (APAP) as a model compound because this drug is known to induce covalent binding to proteins when bioactivated by cytochromes P450 to its reactive N-acetyl-p-benzoquinoneimine metabolite. First, by microsomal incubations of APAP in presence of CPF and/or intact albumin, in vitro reference adducts were generated to determine the mass spectrometric characteristics of the expected CPF adducts and to confirm their formation on pronase E digestion of the alkylated protein. When applying this methodology to albumin isolated from blood of patients exposed to APAP, we were indeed able to detect the corresponding CPF adducts. Therefore, this strategy could be seen as a potential biomonitoring tool to detect in vivo reactive intermediates of drugs and drug candidates, e.g., in the preclinical and clinical development phase.
Although much effort has been made in the development of predictive animal models useful for the early assessment of toxicity of drugs and drug candidates, the prediction of drug toxicity in humans stays difficult. Whereas some adverse drug reactions (ADRs) can be predicted from preclinical safety studies, others are idiosyncratic in nature and only show up after the drug is already introduced on the market. These idiosyncratic types of drug reactions can lead to severe, and, in some cases, fatal toxicities in several organs, in particular the liver, skin, and blood (Park et al., 2005; Smith and Schmid, 2006).
Even though the underlying mechanisms of most ADRs are as yet poorly understood, formation of reactive metabolites is considered to be a major trigger in the cascade of events leading to these adverse events (Williams et al., 2002). Drugs can be bioactivated both by phase I and phase II enzymes to reactive electrophilic intermediates, which subsequently react with nucleophilic sites in macromolecules to form covalent adducts to proteins (Evans et al., 2004; Zhou et al., 2005). Covalent binding to proteins with subsequent inactivation of enzymes and/or disruption of cellular signaling processes are events that are thought to be related to the onset of ADRs (Zhou et al., 2005). By serving as haptens, drug-protein adducts may also trigger the autoimmune reactions, which are often observed in case of idiosyncratic drug reactions (IDRs) (Uetrecht, 1999; Park et al., 2000).
As reviewed by Caldwell and Yan (2006) and Zhou (2003), different methodologies are used for the detection of adducts resulting from formation of reactive intermediates. Briefly, these methods involve in silico screening of potentially toxic motives in molecules, the use of small nucleophilic trapping agents, followed by mass spectrometric analysis of adducts formed in vitro and mechanism-based inhibition of cytochrome P450. An estimation of the levels of total covalent binding to proteins, in vitro and/or in vivo, can eventually be obtained by using radiolabeled drugs in animal experiments. However, because extrapolation of animal data to evaluate potential risks in humans stays complicated (Olson et al., 2000; Smith and Schmid, 2006), there is still a need for sensitive and selective methods for the assessment of covalent binding to proteins in vivo in humans. In the present study, a recently developed liquid chromatography/tandem mass spectrometry (LC/MS/MS) methodology will be applied to monitor covalent binding of acetaminophen (APAP) to the blood protein albumin.

Scheme of the strategy proposed for the generation and detection of drug adducts to HSA.
The concept of using blood protein adducts as biomarkers of human exposure to electrophilic compounds dates back to the 1970s and was originally applied for the in vivo monitoring of occupational exposures to reactive, potentially genotoxic compounds (van Welie et al., 1992; Tornqvist et al., 2002). Consistently, adducts to human serum albumin (HSA) have been found in populations exposed to several environmental contaminants (Sherson et al., 1990; Wild et al., 1990; Omland et al., 1994; Rappaport et al., 2005).
The aim of the present study is to evaluate the applicability of a recently developed LC/MS/MS methodology for the in vivo biomonitoring of reactive drug metabolites to the blood protein albumin. This methodology, which has been applied successfully for the biomonitoring of exposure of humans to chemical warfare agents (Noort et al., 1999, 2002), is based on the digestion of albumin by pronase E and subsequent selective detection of covalent adducts to the tripeptide cysteine34-proline-phenylalanine (CPF) by LC/MS/MS. Cysteine34 is the only free thiol group in HSA and is capable of reacting with electrophiles. In this work, we evaluated the applicability of this methodology for the monitoring of reactive drug metabolites using APAP as model compound. At therapeutic doses, APAP is primarily metabolized by phase II enzymes to stable glucuronic acid and sulfate conjugates. A small proportion of the drug is bioactivated by cytochromes P450 to a reactive N-acetyl-p-benzoquinoneimine (NAPQI) intermediate that under normal conditions is detoxified by conjugation to glutathione (GSH) (Gibson et al., 1996; Zhou et al., 1996; Qiu et al., 1998). When taken in overdoses, the high levels of NAPQI produced will deplete the GSH stores, resulting in strongly increased covalent binding to liver proteins, oxidative stress, and ultimately to severe hepatotoxicity (Bessems and Vermeulen, 2001).
We propose a general strategy that consists of the biosynthesis of reference adducts to CPF and albumin to determine the mass spectrometric characteristics of the CPF adducts and to determine whether pronase E treatment of alkylated albumin is able to generate the corresponding CPF adducts (Fig. 1). The analytical procedure was subsequently applied for the measurement of adducts in the blood of humans exposed to high doses of APAP.
Materials and Methods
Materials. Pronase E (protease from Streptomyces griseus, type XIV, 3.4.24.31), GSH (reduced GSH, 98%), and N-acetyl-l-cysteine (NAc, 98%) were purchased from Sigma (Deisenhofen, Germany). NADPH-tetrasodium salt was obtained from AppliChem BioChemica (Darmstadt, Germany). Amicon Ultra-4 (10-kDa molecular mass cutoff) centrifugal filters were purchased from Millipore (Bedford, MA). The HiTrap Blue HP affinity columns (1 ml; prepacked with blue Sepharose high performance, with Cibacron Blue F3G-A as the ligand) and the PD-10 columns (containing 10 ml of Sephadex G 25 material) were obtained from Amersham Biosciences (Uppsala, Sweden). The Acrodisc LC polyvinylidene difluoride filters (0.45 μm, 25 mm) were obtained from Waters Corporation, and the Strata-X columns (33 μm Polymeric Sorbent) were from Phenomenex. β-Naphthoflavone-induced rat liver microsomes (RLM) were prepared according to the standard protocol of our laboratory (Rooseboom et al., 2001). Control human blood was obtained from healthy volunteers, and blood samples from patients overdosed with APAP were provided to us by Dr. D. Touw (Apotheek Haagse Ziekenhuizen, The Hague, The Netherlands). Human plasma exposed to perdeuterated sulfur mustard was used as internal standard and was prepared as described previously (Noort et al., 2004). All the other chemicals were of the highest grade and were obtained from standard providers.
Instrumentation. LC/electrospray (ES)-MS(/MS) analyses were conducted on a Q-TOF hybrid instrument (Micromass, Altrincham, UK) equipped with a standard Z-spray ES interface (Micromass) and an Alliance, type 2690 liquid chromatograph (Waters, Milford, MA). The chromatographic hardware for this system consisted of a precolumn splitter (type Acurate, LC Packings, Amsterdam, The Netherlands), a six-port valve (Valco, Schenkon, Switzerland) with a 50-μl injection loop mounted, and a PepMap C18 column (15 cm × 1mm i.d., 3-μm particles, LC Packings). A gradient of eluents A (H2O with 0.2% formic acid) and B (acetonitrile with 0.2% formic acid) was used to achieve separation, following 100% A (at time 0 min, 0.1 ml/min flow) to 100% A (at 5 min, 0.6 ml/min flow) to 30% A and 70% B (at 60 min, 0.6 ml/min flow). The flow delivered by the liquid chromatograph was split precolumn to allow a flow of approximately 40 μl/min through the column and into the ES-MS interface. The Q-TOF was operated at a cone voltage of 20 to 30 V, using nitrogen as the nebulizer and desolvation gas (at a flow of 20 and 400 l/h, respectively). MS/MS product ion spectra were recorded using collision energy between 20 and 30 eV, with argon as the collision gas (at an indicated pressure of 10–4 mbar).
Standard Tripeptide Assay. Adducts to HSA were analyzed using a slightly modified methodology that has been described previously (Noort et al., 2004). Briefly, human blood was first centrifuged at 3000g to separate plasma from erythrocytes. The obtained plasma (500 μl) was then diluted with 2 ml of buffer A (50 mM KH2PO4, pH 7.00). To control the procedure, the samples were spiked with 50 μl of an internal standard consisting of plasma isolated from human blood exposed to 100 μM of perdeuterated sulfur mustard gas. The pronase E digest of albumin alkylated with perdeuterated sulfur mustard gas is known to produce the characteristic d8-S-(2-hydroxyethylthioethyl)-Cys-Pro-Phe (d8-HETE-CPF) adduct, as has been described in Noort et al. (2004). The samples were then filtered with 0.45-μm Acrodisc filters, and the albumin was subsequently isolated from the filtrate using HiTrap Blue HP affinity columns. These columns were first conditioned with 10 ml of buffer A. The whole sample (2.55 ml) was then applied on the columns and washed with 10 ml of buffer A. Elution took place with 3 ml of buffer B (50 mM KH2PO4 with 1.5 M KCl). The HiTrap columns were regenerated by washing with 10 ml of buffer A. PD-10 columns were used to desalt the obtained albumin fractions. After equilibration of the PD-10 columns with 25 ml of 50 mM NH4HCO3, the samples were applied on the columns (3 ml) and eluted with 3 ml of the same bicarbonate buffer. The digestion procedure of the desalted albumin solution with pronase E was as follows: 100 μl of a freshly prepared pronase E solution (10 mg/ml stock solution in aqueous 50 mM NH4HCO3) was added to 750 μl of the albumin fraction (in 50 mM NH4HCO3). After 2 h of incubation at 37°C, the mixture was passed through molecular mass cutoff filters (10 kDa) under centrifugation at 2772g to remove the enzyme. Under these conditions, pronase E digestion of albumin adducts is completed (Noort et al., 1999). The filtrate was subsequently analyzed by LC/MS/MS. If not analyzed immediately, all the samples were stored at – 20°C until analysis.

Electrospray product ion spectrum of the NAPQI-CPF adduct. The reference NAPQI-CPF adduct was obtained from the reaction of synthetic NAPQI with synthetic tripeptide CPF. The electrospray product ion spectrum of the adduct (MH+ 515.18) was obtained with a collision energy of 24 eV.
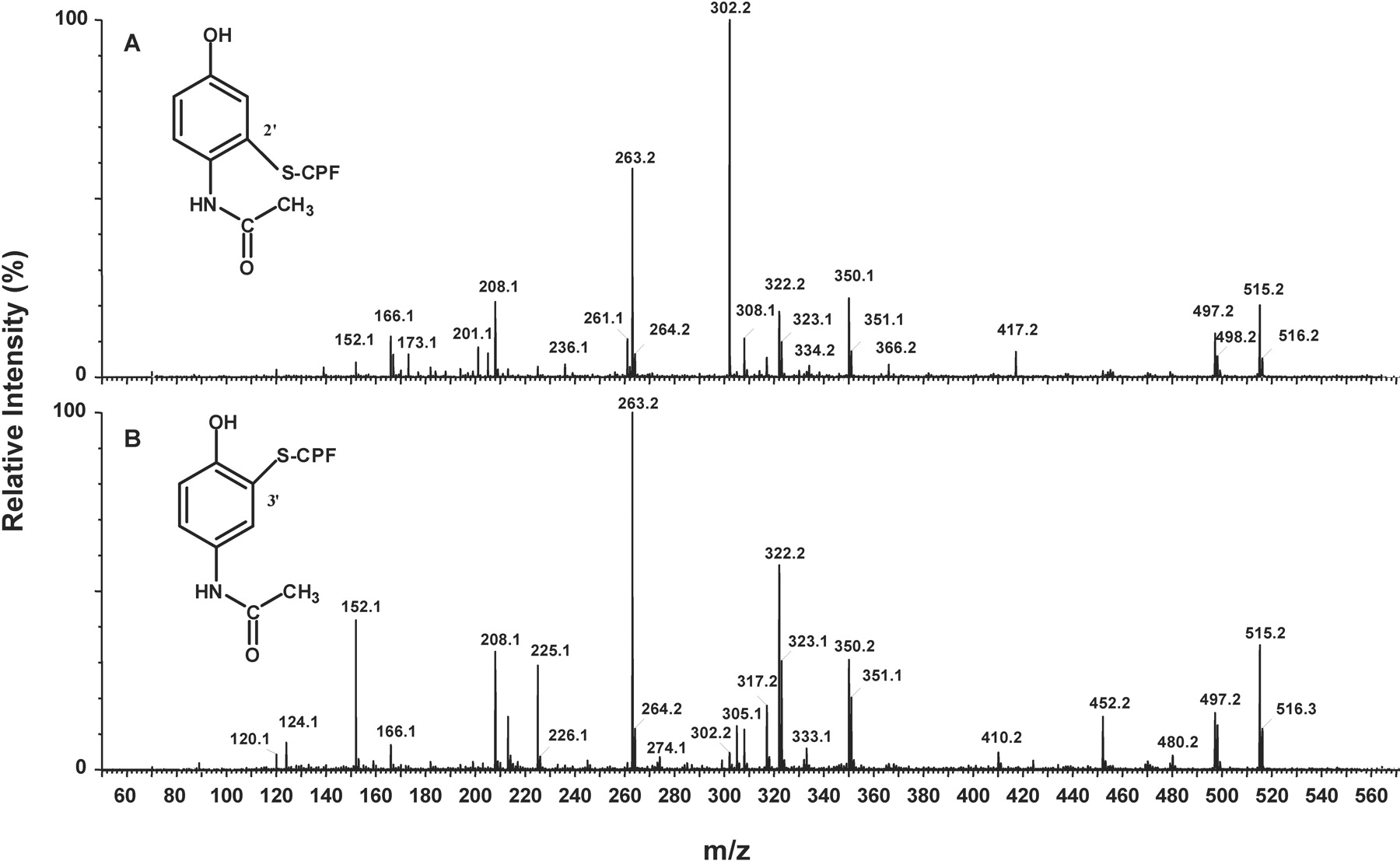
Synthesis and Purification of NAPQI-CPF Adducts. The reactive metabolite of APAP, NAPQI, was synthesized according to a previously described method (Bessems et al., 1996). Briefly, fresh silver oxide was prepared by adding a silver nitrate solution (170 mg in 10 ml of H2O) to a solution of potassium hydroxide (100 mg in 18 ml of H2O). The mixture was left for 15 min on ice to yield the highest amounts of the silver oxide precipitate. After filtration of the solution and three washing steps with acetone, the obtained silver oxide powder was added to a solution of APAP (10 mg in 10 ml of chloroform). This reaction mixture was stirred at room temperature for 1 h to obtain 10 ml of a yellowish NAPQI solution (in chloroform). A portion of this NAPQI solution (2 ml) was filtered, evaporated to dryness by rotaevaporation at room temperature, and further taken up in 10 μl of acetonitrile. This concentrated NAPQI solution was then reacted with 100 μl of synthetic CPF tripeptide solution (stock concentration 2.5 mM in 50 mM NH4HCO3) in a total volume of 1.5 ml of a 30% acetonitrile solution (in bicarbonate buffer). After2hof reaction at 37°C, the sample was measured by LC/MS/MS. LC/MS/MS analysis of the reaction mixture showed the presence of a product having the molecular mass of the expected NAPQI-CPF adduct (Fig. 2). The product ion spectrum of this protonated molecule (m/z 515.18 [MH+]) had characteristic fragments at m/z 497.18 (MH+ – H2O), m/z 350.11 (b2), m/z 322.13 (a2), m/z 263.16 (y2″), m/z 225.07 (a1), m/z 208.06 (a1-NH3), m/z 166.06 (y1″), m/z 152.07 (APAP + H+), and m/z 120.07 (immonium ion of phenylalanine).
To further characterize the NAPQI-CPF product and to enable quantification of NAPQI-CPF in patient samples, a large-scale synthesis was performed. APAP (32 mg) was oxidized to NAPQI in 75 ml of chloroform, as described above, and stirred vigorously for 2 h at room temperature with 25 mg of CPF in 30 ml of 100 mM potassium phosphate buffer, pH 7.4. The resulting water phase was isolated and washed three times with a mixture of chloroform/isopropanol (3:1 v/v) to remove excess NAPQI and lipophilic side products. The water phase was subsequently dried overnight by nitrogen stream. The residue was taken up in 1 ml of 10% acetonitrile and applied to preparative high-performance liquid chromatography (HPLC) to purify NAPQI-CPF adducts.
The preparative HPLC consisted of a Luna C18 column (250 × 10 mm, 5-μm particles) eluted at a flow rate of 2 ml/min. A gradient of eluents A (1% acetonitrile/0.2% formic acid/98.8% water) and B (99% acetonitrile/0.2% formic acid/0.8% water) was used to achieve separation of analytes. The gradient used started at 15% B (0 min) and was followed by a linear increase to 25% B (50 min). Peaks absorbing at 254 nm were collected and screened for presence of NAPQI-CPF by LC/MS. Two peaks at 21.9 (minor, 5%) and 23.6 min (major, 95%) were shown to contain NAPQI-CPF. The fractions containing purified NAPQI-CPF were pooled, dried by nitrogen stream, and taken up in 500 μl of deuterium oxide. The 1H NMR spectrum was recorded on a Bruker MSL 400 system operating at 376.43 Hz. The 1H NMR spectrum of the major NAPQI-CPF isomer was consistent with an NAPQI-CPF adduct, although protons could not be assigned individually because of the complexity and overlap of the 1H NMR signals. Because aromatic protons of the phenylalanine residue interfere with the signals of the APAP residue, the position of the thioether bond could not be assigned. 1H NMR spectrum: 6.8 to 7.6 ppm (aromatic protons; multiplet; 8H), 3.8 to 4.5 ppm (methine protons of phenylalanine, cysteine, and proline; multiplet; 3H), 2.8 to 3.55 ppm (methylene protons of phenylalanine and cysteine; four methylene protons of proline; multiplet; 8H), and 1.65 to 2.3 ppm (two methylene protons of proline and acetyl protons of APAP; multiplet; 5H). The concentration of the minor NAPQI-CPF isomer was too low to obtain a good 1H NMR spectrum.
To calibrate the concentration of NAPQI-CPF solution, difluoroacetic acid (final concentration, 720 μM) was added to the solution of NAPQI-CPF and analyzed by 1H NMR. Difluoroacetic acid was selected as internal standard because its signals (triplet at 5.82 ppm, 2JFH 51 Hz) do not interfere with the signals of NAPQI-CPF. By integrating the signals of aromatic protons of NAPQI-CPF ([8H]) and that of difluoroacetic acid ([1H]), the concentration of the NAPQI-CPF solution was estimated to be 650 μM. This solution was used for the quantification of NAPQI-CPF adducts formed by pronase E treatment of patient samples.
Incubation of Synthetic NAPQI in Human Plasma. The synthetic NAPQI solution described above was used to expose human plasma to the reactive alkylating compound. In these experiments, 3 ml of the NAPQI solution (in chloroform) was evaporated to dryness by rotaevaporation at room temperature and further taken up in 10 μl of acetonitrile. The concentrated NAPQI solution was then reacted with 2 ml of human plasma for 2 h at 37°C. Subsequently, 500 μl of that incubation mixture was diluted with 2 ml of buffer A and spiked with 50 μl of the internal standard. The sample was consistently passed through the Acrodisc filter, and the albumin was isolated on the HiTrap column as described above. The albumin fraction was then desalted on PD-10 columns, and 750 μl of that filtrate was treated with pronase E for 2 h at 37°C. After a centrifugation step with the molecular mass cutoff filters, the filtrate was analyzed by LC/MS/MS.
Microsomal Incubations of APAP in Presence of Synthetic CPF. Microsomal incubations of APAP and CPF had a final volume of 500 μl and were conducted at 37°C in a heated shaking water bath. The incubation occurred in a 100 mM KH2PO4 buffer, pH 7.4, and the mixture was composed as follows (in final concentrations): 1 mM APAP, β-naphtoflavone-induced RLM (2 mg protein/ml), and 1 mM synthetic CPF tripeptide solution. After 5 min of preincubation, the cofactor NADP(H) (final concentration, 2 mM) was added to the mixture, and the samples were incubated for 1 h at 37°C. After the incubation time, the samples were placed on ice, and 200 μl of 2.00 M HClO4 was added to stop the incubations and precipitate proteins. The samples were vortexed and kept on ice for an additional 5 min. The tubes were subsequently centrifuged for 15 min at 4000 rpm, and a 400-μl aliquot of the supernatant was neutralized by the addition of an equal volume of 1.00 M K2HPO4. The samples were vortexed and centrifuged again for 15 min at 4000 rpm, and the supernatant was analyzed by LC/MS/MS. An incubation performed without APAP was processed in parallel and served as control.
Microsomal Incubations of APAP in Presence of Human Plasma. Microsomal incubations of APAP in human plasma were performed similarly as described above. Briefly, 75 μl of APAP (20 mM stock in H2O) and 150 μlof RLM (10 mg protein/ml stock) were added to 500 μl of human plasma. After 5 min of preincubation at 37°C, 150 μl of NADP(H) (10 mM stock in 100 mM K-P buffer, pH 7.4) was added to the incubation mixture. The mixture was incubated for 1 h at 37°C. After the incubation time, 2 ml of buffer A and 50 μl of the internal standard were added to the samples. The albumin isolation, pronase E digestion, and peptide filtration steps were performed similar to the procedure described for the exposure of human plasma to synthetic NAPQI. The samples were subsequently analyzed by LC/MS/MS. Incubation performed without substrate served as control.
Analysis of Human Serum Samples. Consistently, 500 μl of human serum of patients exposed to high levels of APAP was diluted with 2 ml of buffer A, and the samples were spiked with 50 μl of the internal standard. The rest of the procedure was similar to that described earlier. Blood from healthy volunteers, not being exposed to either APAP or NAc, was taken as control and processed in parallel.
Initial measurements of the patient's blood samples revealed a significant background signal interfering with the expected NAPQI-CPF adducts. Therefore, to increase the sensitivity in the albumin adduct detection, the protocol was slightly adapted by including a solid-phase extraction step in the procedure. The human serum samples were analyzed with the modified protocol as follows. After isolation and desalting of albumin as described above, 300 μlof a fresh pronase E solution (10 mg/ml) was added to approximately 2 ml of the albumin solution. After2hof incubation at 37°C, the samples were centrifuged at 2772g to eliminate the excess of enzyme. The freshly digested samples were acidified with trifluoroacetic acid (TFA) (final concentration 0.1% TFA), and Strata X columns were used for the solid-phase extraction procedure. The columns were first activated with 10 ml of methanol and then equilibrated with 10 ml of 100% H2O with 0.1% TFA. The samples were applied on the column and subsequently eluted with 2-ml fractions of 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100% acetonitrile solutions containing 0.1% TFA. These different fractions were collected, freeze-dried, taken up in 120 μl of 100% H2O with 0.2% formic acid, and analyzed by LC/MS/MS. The NAPQI-CPF adducts were eluted by the 30% acetonitrile/0.1% TFA fraction.
To quantify NAPQI-CPF adducts formed by pronase E treatment of albumin, the albumin fractions isolated from the plasma samples were also spiked with 65 nmol of purified synthetic NAPQI-CPF (major isomer) before the treatment with pronase E. By measuring the increase in the ratio of the peak area of NAPQI-CPF with that of the internal standard (d8-HETE-CPF), the amount of NAPQI-CPF in the nonspiked albumin fraction was estimated.
To analyze small molecular weight adducts, the eluates obtained from the application of the serum samples on the HiTrap columns were collected, filtered with molecular mass cutoff filters (10 kDa) under centrifugation at 2772g, and analyzed by LC/MS/MS.
Results
Incubation of Synthetic NAPQI with Human Plasma. To assess whether NAPQI is also reactive toward the free cysteine34 residue of HSA and whether the pronase E digest of alkylated albumin is leading to the formation of NAPQI-CPF, human plasma was incubated in vitro with freshly synthesized NAPQI. Full MS analysis was used to screen the fragments obtained after fragmentation of the selected molecular ion with an m/z of 515.2. The reconstructed ion chromatogram of the summed ions corresponding to specific fragments of the synthetic adduct revealed the presence of two NAPQI-CPF adducts in the reaction mixture (peaks 1 and 2 in Fig. 3A). Their product ion spectra were comparable with that of our reference adduct. Only slight differences in the fragmentation pattern of the two peaks were observed, suggesting the formation of two regioisomeric NAPQI-CPF adducts.
Incubations of APAP with RLM. Next to the chemical synthesis of the albumin and CPF adducts of NAPQI, it was also investigated whether in vitro incubations of APAP in presence of albumin or CPF produced the same adducts. Therefore, we used βNF-induced RLM to bioactivate APAP to NAPQI in a biological system. By performing incubations using synthetic CPF as trapping agent for NAPQI, two NAPQI-CPF adducts were found by LC/MS/MS analysis (Fig. 3B).
When APAP was incubated with RLM in presence of human plasma, two NAPQI-CPF adduct isomers were observed after pronase E digestion of the albumin fraction. The adducts had the same characteristic differences in their product ion spectra as observed in the products formed by synthetic NAPQI (Fig. 3C).
Analysis of Human Serum Samples. The developed methodology was applied to analyze blood of patients exposed to high levels of APAP. Three patients were selected with the characteristics presented in Table 1. Patient TOX 444 suffered from severe hepatotoxicity as indicated by increased plasma aspartate transaminase and alanine aminotransferase levels. The two other patients (TOX 438 and TOX 440) had normal plasma transaminase levels and were therefore considered to be in a nonhepatotoxic condition. Initially, the serum samples of these patients were analyzed with the standard tripeptide assay as described in Noort et al. (2004). However, by this assay no NAPQI-CPF adducts were observed in the sera of patients TOX 438 and TOX 440 as a result of background interference, whereas two weak NAPQI-CPF signals were found in the serum of patient TOX 444 (data not shown). By using the solid-phase extraction step in the procedure, it was found that the NAPQI-CPF peaks eluted in the 30% acetonitrile/0.1% TFA fraction. The signal-to-noise ratio of the NAPQI-CPF signals was significantly increased using this clean-up step. Two NAPQI-CPF adducts were observed in the sera of patient TOX 444, and signals corresponding to the major NAPQI-CPF adduct were now also observed in the sera of patients TOX 438 and TOX 440. Based on the peak areas of the summed ion current chromatograms, the level of the major NAPQI-CPF adduct was about 10-fold higher in serum of TOX 444 when compared with that in serum of TOX 438 and 440. The product ion spectra of these adducts were identical to those obtained in our previous in vitro experiments. No NAPQI-CPF adducts were present in the control subjects (Fig. 5).
Characteristics of the patients overdosed with acetaminophen
The table includes the ingested dose of APAP, the time after APAP intake, the blood concentration of APAP in time, whether the patient was treated with NAc, and the levels of hepatic enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in time.

Analysis of NAPQI-CPF adducts (MH+ 515.2) generated in different in vitro systems. Ion chromatograms of summed ions: m/z 152.1, 208.1, 225.1, 322.1, 350.2 in a pronase digest of alkylated HSA isolated from human plasma exposed to synthetic NAPQI (A), an APAP microsomal incubation performed with synthetic CPF (B), a pronase digest of alkylated HSA isolated from an APAP microsomal incubation performed in human plasma (C).
By spiking the isolated albumin fractions of patient TOX 444 with 65 nmol of synthetic NAPQI-CPF, before pronase E treatment, the amount of NAPQI-CPF in the pronase E digest was quantified. By comparing the peak areas of NAPQI-CPF in samples of patient TOX 444 with and without spiking with synthetic NAPQI-CPF, the amount of NAPQI-CPF in the unspiked sample was estimated to be 35 ± 15 pmol/ml serum. Based on the 10-fold lower peak areas in patient samples TOX 438 and 440 (Fig. 5), the levels of NAPQI-CPF in these serum samples are estimated to be approximately 3 to 4 pmol/ml serum.
The eluates obtained during the albumin isolation step were also screened for the presence of other NAPQI-derived adducts. No GSH conjugates were observed in the human plasma samples. However, very high amounts of NAPQI-cysteine (NAPQI-Cys) and NAPQI-N-acetyl cysteine (NAPQI-NAc) adducts were found in the sera of these patients (Fig. 6). Their specific fragmentation pattern confirmed the identity of these adducts of which the characteristics are presented in Table 2.
Characterization of the NAPQI-Cys and NAPQI-NAc adducts
Characteristics of the electrospray product ion spectra of the NAPQI-Cys and of the NAPQI-NAc adducts detected in the human serum samples. Product ion spectra were obtained with a collision energy of 15 eV.
Interestingly, another high signal was found in the pronase E digest of the isolated albumin fraction of patient TOX 444, suggesting that another albumin adduct is formed in vivo. The ion chromatogram of the protonated molecule (m/z 655.4 [MH+]) is depicted in Fig. 7A. The product ion spectrum of this ion showed characteristic signals at m/z 637.4 (MH+ – H2O), m/z 527.3 (y3″), m/z 490.3 (b3), m/z 393.2 (b2), m/z 375.2 (b2-H2O), m/z 347.2 (a2-H2O), m/z 263.2 (y2″), m/z 230.1 (b2-NAc), m/z 162.1 [NAc-H]+, and m/z 101.1 (a1). This fragmentation pattern is consistent with the glutamine-CPF (QCPF) tetrapeptide, from which the cysteine34 residue formed a mixed disulfide with N-acetyl cysteine (Fig. 7B). This adduct was also found in the serum of patient TOX 438 but was absent in patient TOX 440. Because the formation of a tetrapeptide instead of the CPF tripeptide was surprising, we checked the enzymatic activity of pronase E by analyzing the internal standard that was systematically added to the serum samples. The expected d8-HETE-CPF tripeptide adduct was consistently found in the samples, thereby confirming a correct activity of the protease (data not shown). The human samples were also screened for the possible formation of NAPQI-QCPF adducts, but no signals corresponding to these adducts were observed, suggesting a complete digestion toward NAPQI-CPF adducts. Eventually, the levels of the different adducts analyzed for each patient were found to be constant in time.
Table 3 summarizes the different adducts identified in the human serum samples and gives a semiquantitative overview of their relative concentrations.
Overview of the adducts detected in the human serum samples
Levels of analytes are ranked as very abundant (++++), abundant (+++), average (++), present (+), and absent (-). Because levels are based on peak areas of summed ion chromatograms of each analyte, only levels from the same column should be compared.
Discussion
Predicting the potential of drugs and drug candidates to lead to adverse drug reactions in humans via electrophilic reactive intermediates is still a difficult and speculative task. Whereas the current way to assess this issue is usually to combine in vitro data on reactive intermediate formation with protein covalent binding studies in vivo in animals with radiolabeled drugs, extrapolation of these data to assess potential risks for humans remains complicated (Caldwell and Yan, 2006; Smith and Schmid, 2006). In this study, we evaluated the use of albumin adducts as biomarkers of bioactivation of drugs to reactive metabolites in vivo in humans. An overall strategy for the generation and analysis of those adducts is proposed. The methodology used is based on LC/MS/MS analysis of covalent adducts to free cysteine34 residue of HSA by detecting an alkylated CPF tripeptide obtained after pronase E digestion of the protein. APAP was chosen as model compound to study this concept (Bessems and Vermeulen, 2001; James et al., 2003).

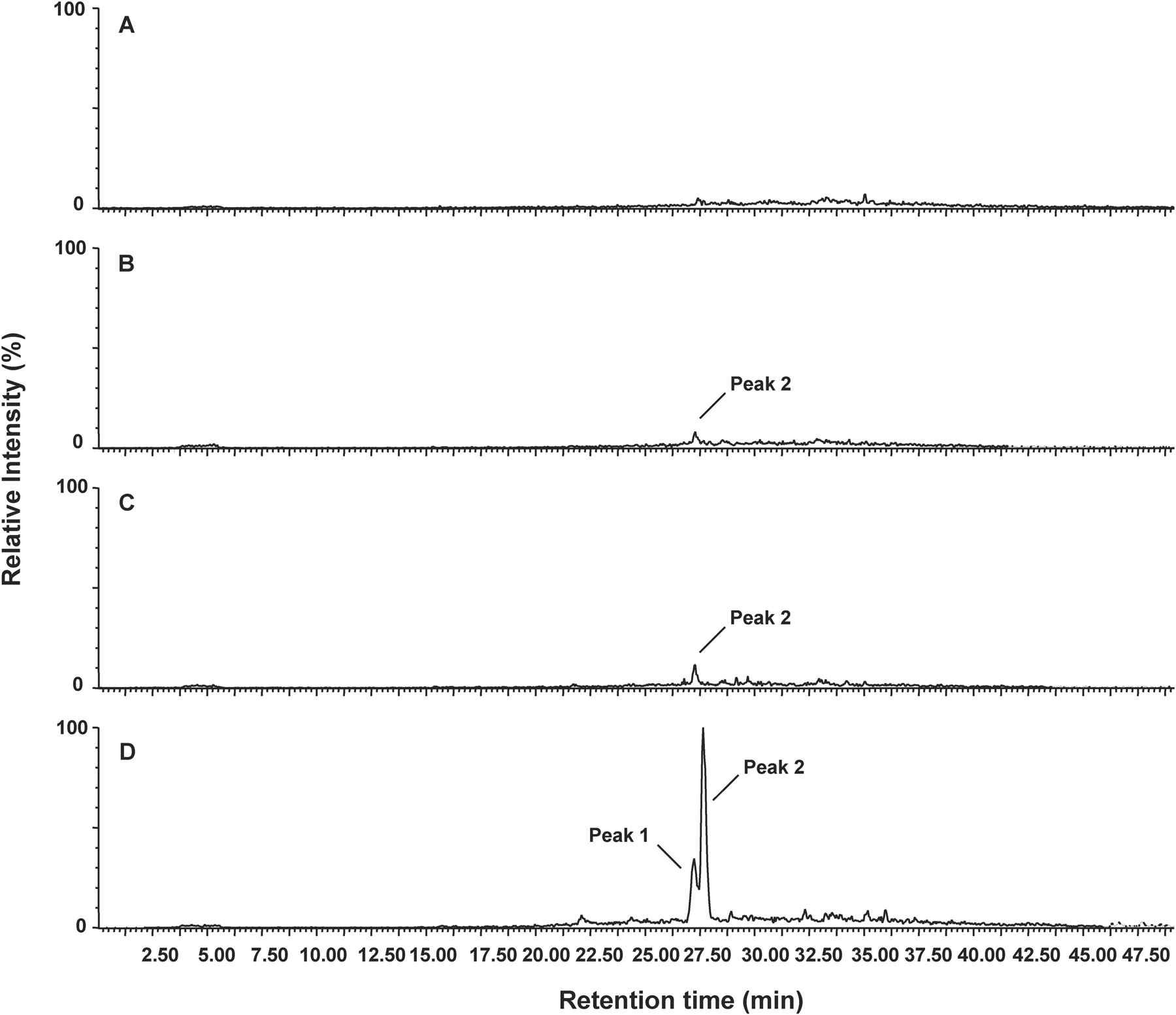
LC/MS/MS analysis of NAPQI-CPF adducts in human serum samples (MH+ 515.2). Ion chromatograms of summed ions: m/z 152.1, 208.1, 225.1, 322.2, 323.2, and 351.2 in the control subject (A), patient TOX 438 (B), patient TOX 440 (C), and patient TOX 444 (D). The analyses of the 23-, 4-, and 25-h time point samples of patients TOX 438, 440, and 444, respectively, are represented here.

Analysis of NAPQI-NAc and NAPQI-Cys adducts in the human serum samples. LC/MS/MS analysis of the NAPQI-Cys adduct (MH+ 271.1; ion chromatogram of summed ions: m/z 140.0, 182.1) (A) and the NAPQI-NAc adduct (MH+ 313.1; ion chromatogram of summed ions: m/z 208.1, 271.1) (B) with proposed structures, analyzed in the 4-h time point sample of patient TOX 440.
We have shown that microsomal incubations were able to generate the same albumin adducts as those obtained with synthetic NAPQI (Fig. 3), indicating that the biosynthesis of reference adducts is possible. Therefore, this biosynthetic approach allows the development of sensitive and selective analytical methods for the detection of CPF adducts without the requirement for chemical synthesis of reference adducts. The NAPQI-CPF adducts observed had slightly different product ion spectra and consequently suggested the formation of two isomers. Based on literature, we assume that the major peak is most likely the 3′-NAPQI-S-CPF adduct because several studies have shown that conjugation of thiols to NAPQI predominantly takes place at the 3′ position of NAPQI (Hoffmann et al., 1985; Axworthy et al., 1988; Pumford et al., 1997; Chen et al., 1999). As the minor NAPQI-CPF adduct, two different products can be considered: a thioether ipso adduct and a 2′-NAPQI-S-CPF adduct. Chen et al. (1999) showed that the ipso adduct was formed at slightly higher levels than the 2′-isomer after reaction NAPQI with GSH at pH 6. However, the ipso adduct was shown to be highly unstable at a lower pH, with a half-life of 0.5 min at pH 4. Because of the lengthy procedure, involving albumin isolation and pronase E digestion, and the fact that the solid-phase extraction was performed at acid pH (∼pH 2), it seems unlikely the minor NAPQI-CPF adduct will correspond to the ipso adduct. Therefore, we propose that the minor NAPQI-CPF adduct corresponds to the 2′-regioisomer.
When applying the developed methodology to blood samples from patients exposed to high doses of APAP, the major NAPQI-CPF isomer was observed in all the patients (Fig. 5). The patient with severe hepatotoxicity, TOX 444, showed an approximately 10-fold higher level of NAPQI-CPF adduct when compared with the patients without hepatotoxicity. The minor NAPQI-CPF isomer could only be observed in the blood sample of patient TOX 444. To our best knowledge, this is the first time that two NAPQI-HSA adduct regioisomers have been observed in vivo in humans. The levels of NAPQI-CPF adduct present in the serum of patient 444 were estimated to be about 35 pmol/ml of serum. It has been reported previously that significantly higher levels of NAPQI-protein adducts can be found in serum in patients showing severe hepatotoxicity in comparison with serum of patients without or with low hepatotoxicity (Muldrew et al., 2002; James et al., 2006). However, because a different methodology was used in these studies, measuring total NAPQI-Cys adducts after complete hydrolysis of whole serum, the levels of our NAPQI-CPF adducts were not compared quantitatively with the adduct levels reported previously.

Analysis of the QCPF-NAc adduct in the human serum samples. LC/MS/MS analysis of the QCPF-NAc adduct (MH+ 655.4) in a pronase digest of alkylated HSA isolated from the 25-h time point sample of patient TOX 444: ion chromatogram of m/z 655.4 (A) and its product ion spectra with proposed structure (B).
Besides the NAPQI-CPF adducts, very high levels of NAPQI-Cys and NAPQI-NAc adducts were found in the sera of all the patients. These adducts probably originate either from the direct trapping of NAPQI by the antidote N-acetyl-cysteine and/or from the catabolism of corresponding GSH adducts (Commandeur et al., 1995; Bessems and Vermeulen, 2001). However, the fact that patient 440, who did not receive NAc, forms equal amounts of NAPQI-NAc suggests that the major amount may be derived from GSH adduct catabolism. The formation of the QCPF-NAc adduct can be explained by mixed disulfide formation between the cysteine thiol of HSA and NAc, as has been shown in vivo in the plasma of rats treated with NAc (Harada et al., 2002). Because NAPQI is a strong oxidizing agent (Gibson et al., 1996) and the formation of mixed disulfides is a known indicator of oxidation (Eaton, 2006), the QCPF-NAc adduct could therefore be considered as a marker of oxidative stress. However, because human blood samples from patients exposed to NAc and not to APAP are not available, the precise impact of NAPQI on QCPF-NAc adduct formation could not be directly assessed.
From the present results, it is clear that, although the doses of APAP were extremely high, the levels of NAPQI-HSA adducts in the human samples were unexpectedly low. One explanation may be the extensive mixed disulfide formation resulting from the oxidative properties of NAPQI that may have prevented the formation of NAPQI-CPF adducts by blocking the free cysteine34-thiol group of HSA. The fact that lower amounts of NAPQI-CPF adducts were observed in the blood of patient TOX 438, who ingested similar amounts of APAP than TOX 440 but was treated with NAc, might support this hypothesis. Another explanation is that alkylation is taking place to prealbumin in the liver and that the NAPQI-CPF adducts found in plasma result from the leakage of albumin adducts into the bloodstream in cases of overt liver damage. This is consistent with the observation that much higher levels of NAPQI-CPF adducts were found in patient TOX 444, which showed high leakage of transaminases, compared with the adduct levels detected in patients TOX 438 and 440. Figure 8 summarizes the proposed interaction between NAPQI and the various trapping reactions involved.
Previous methods that have been used to investigate covalent binding of APAP to proteins in vivo include several immunoassays (Roberts et al., 1987; Bartolone et al., 1988; Matthews et al., 1996; James et al., 2001) and the use of radiolabeled APAP (Axworthy et al., 1988; Zhou et al., 1996; Qiu et al., 1998). Still, these methods often lack the necessary sensitivity when concerning human serum samples (Zhou, 2003). More recently, an HPLC/electrochemical detection methodology was developed by Muldrew et al. (2002) to quantify covalent protein binding of APAP. The methodology involves the dialysis of serum protein samples, subsequent digestion with protease, and the measurement of the NAPQI-Cys adducts formed. Although the sensitivity of the HPLC/electrochemical detection method was increased compared with the previously mentioned technologies, it remains a long and labor-intensive procedure. Comparatively, our LC/MS/MS methodology is quicker, sensitive, particularly selective, and can provide precise structural information on the adducts formed to HSA as proven by the detection of two regioisomeric NAPQI adducts. This method also proved valuable to quantify the amounts of NAPQI-CPF adducts measured in the human serum samples. However, when synthesis of the reference drug-CPF adduct is not possible, this methodology can already as such be applied for the retrospective and/or relative comparison of drug-albumin adduct levels between individuals. Eventually, the possibility of scanning for neutral losses corresponding to “non–drug-related” fragments of drug-CPF adducts (e.g., the y2″ fragment corresponding to the PF part of the drug-CPF adduct) would allow the detection of novel, yet uncharacterized, drug-protein adducts.

General scheme proposing the different trapping reactions involved in the formation of the adducts detected in the human serum samples.
In summary, we propose a generic strategy to assess the potential of drugs to be bioactivated to reactive electrophilic metabolites and to covalently bind to proteins in vivo (Fig. 1). The strategy consists first in the biosynthesis of reference drug-albumin adducts for method development purposes. This biosynthetic approach is of particular importance because the synthesis of reactive metabolites of drugs is generally not feasible. Second, the developed analytical methodology is applied in the “in vivo approach” where serum samples of patients exposed to the drug are analyzed for the presence of similar albumin adducts. Smaller adducts, such as N-acetyl cysteine and cysteine adducts, can be measured as well, hereby further confirming the bioactivation potential of the drug in vivo. Although many ADRs seem not to be directly related to the concentration of the parent drug, it is likely that the onset of ADRs is related to the levels of reactive metabolites formed in vivo. Consequently, information on blood levels of “drug-protein adducts” will give an indication of the potential of a drug to be bioactivated to reactive electrophilic metabolites in vivo, and consequently on potential ADRs. Because HSA has a half-life of approximately 20 days, these drug-HSA adducts may accumulate in time. This could be of interest because IDRs usually have a delayed onset of occurrence and seem to take place essentially in patients taking a drug in relatively high doses for a longer period of time. In this perspective, this strategy may also be seen as an in vivo dosimetry methodology to assess levels of covalent binding to proteins (Yang et al., 2006). Consequently, this technology could constitute a potential biomonitoring tool that could improve the risk assessment of ADRs and IDRs of novel drugs and/or drug candidates.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.014233.
-
ABBREVIATIONS: ADR, adverse drug reaction; IDR, idiosyncratic drug reaction; LC/MS/MS, liquid chromatography/tandem mass spectrometry; APAP, acetaminophen; HSA, human serum albumin; CPF, cysteine-proline-phenylalanine; NAPQI, N-acetyl-p-benzoquinoneimine; GSH, glutathione; NAc, N-acetyl-l-cysteine; RLM, rat liver microsome(s); ES, electrospray; d8-HETE-CPF, d8-S-(2-hydroxyethylthioethyl)-Cys-Pro-Phe; HPLC, high-performance liquid chromatography; TFA, trifluoroacetic acid; NAPQI-Cys, NAPQI-cysteine; NAPQI-NAc, NAPQI-N-acetyl cysteine; QCPF, glutamine-CPF.
- Received December 7, 2006.
- Accepted May 16, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}