


Abstract
To determine the effect of intestinal cytochrome P450 (P450) enzymes on the bioavailability of oral drugs, we have examined the metabolism of nifedipine, an antihypertensive drug and a model substrate of CYP3A4, in mouse models having deficient expression of the NADPH-cytochrome P450 reductase. Initial studies were performed on Cpr-low (CL) mice, which have substantial decreases in Cpr expression in all tissues examined, including the small intestine. In CL mice, area under the concentration-time curve (AUC) values for blood nifedipine after intraperitoneal and oral dosing were 1.8- and 4.0-fold, respectively, higher than in wild-type mice, despite increased expression of multiple P450 enzymes in both liver and intestine. The greater extent of the increase in the AUC value for oral than for intraperitoneal nifedipine suggested that intestinal P450s influence the bioavailability of oral nifedipine, a notion supported by results from further studies on LCN and CL-LCN mice. The LCN mice, which have liver-specific Cpr deletion, had 6.9-fold higher AUC values and 2.2-fold higher Cmax values for blood nifedipine than did wild-type mice after oral nifedipine, consistent with the critical role of hepatic P450s in systemic nifedipine clearance. However, in the CL-LCN mice, which have global decreases in Cpr expression in all tissues examined and Cpr deletion in the liver, AUC and Cmax values for oral nifedipine were, respectively, 2.2- and 1.8-fold higher than in LCN mice, confirming the fact that P450-catalyzed metabolism in the small intestine, the portal-of-entry organ for oral drugs, plays an important role in the first-pass clearance of oral nifedipine.
The small intestine is the initial site of metabolism of ingested xenobiotics, including therapeutic drugs, through reactions catalyzed primarily by the cytochrome P450 (P450) oxidative xenobiotic-metabolizing enzyme system. P450 is a superfamily of heme-containing monooxygenases (Nelson et al., 2004), many of which are expressed in the small intestine (Kaminsky and Fasco, 1991; Kaminsky and Zhang, 2003) and are active in the bioactivation or detoxification of numerous toxic chemicals, carcinogens, and therapeutic drugs. It has been proposed that the expression levels and the activities of P450 enzymes in the small intestine directly affect the bioavailability of many drugs (Suzuki and Sugiyama, 2000; Doherty and Charman, 2002; Ding and Kaminsky, 2003); however, few studies have examined the relative contributions of liver and small intestine to systemic bioavailability of oral drugs, mainly because of the difficulty of distinguishing between hepatic and intestinal first-pass metabolism. The expression of small intestinal P450 enzymes is regulated by exposure to dietary and xenobiotic compounds (Kaminsky and Fasco, 1991; Zhang et al., 1996, 2003), as well as by pathologic conditions such as inflammation (Kalitsky-Szirtes et al., 2004; Xu et al., 2006). Therefore, knowledge of whether the small intestine has a significant influence on bioavailability of an oral drug will facilitate development of better therapeutic strategies that enhance efficacy and minimize toxicity in patients.
Microsomal P450 enzymes require a redox partner, the NADPH-cytochrome P450 reductase (CPR or POR), for their monooxygenase function (Black and Coon, 1987; Strobel et al., 1995). Several mouse models targeting the Cpr locus have been developed, including the “Cpr-low” (CL) mouse (Wu et al., 2005), the “liver-Cpr-null” (LCN) mouse (Gu et al., 2003; Henderson et al., 2003), and the “Cpr-low and liver-Cpr-null” (CL-LCN) mouse (Gu et al., 2007). In the CL mice, CPR expression is decreased by >70% in all tissues examined, including olfactory mucosa, adrenal gland, brain, testis, ovary, lung, kidney, liver, and heart (Wu et al., 2005). The decreased CPR expression is accompanied by compensatory increases in hepatic as well as renal microsomal P450 content, but also by reductions in liver microsomal metabolism of acetaminophen and testosterone (Wu et al., 2005). In the LCN mouse, Cpr expression is abolished in essentially all hepatocytes, but it is normal in other tissues (Gu et al., 2003). In the CL-LCN mouse, which combines the phenotypes of the CL and LCN mice, Cpr expression is absent in hepatocytes of adult mice, whereas CPR levels are substantially decreased in other tissues (Gu et al., 2007). These mouse models have made it possible to determine the relative contributions of liver and extrahepatic tissue P450 enzymes in the disposition and bioactivation of xenobiotic compounds. For example, it was found that the contribution of hepatic P450 to toxicity of acetaminophen (given intraperitoneally) under overdose conditions differed for various target organs. Whereas toxicity of acetaminophen in the nasal mucosa was not dependent on hepatic CPR- and P450-catalyzed metabolic activation, toxicities in the lung, kidney, and lateral nasal glands were at least partly prevented by the loss of hepatic acetaminophen metabolism in the LCN mice (Gu et al., 2005). Hepatic CPR/P450 was also found to play significant roles in systemic disposition of cyclophosphamide (Pass et al., 2005) but apparently not in the clearance of diclofenac and doxorubicin (Henderson et al., 2006). A role for extrahepatic tissue P450 enzymes in the in vivo disposition of cyclophosphamide was subsequently demonstrated through comparison of the pharmacokinetics of cyclophosphamide metabolism between the LCN and CL-LCN mice (Gu et al., 2007). It should be noted that none of these studies compared the impact of CPR deficiency on the disposition of intraperitoneally administered drugs with the impact on orally administered drugs; for oral drugs, the small intestine is expected to play a greater role in first-pass metabolism than for drugs given intraperitoneally (Grundy et al., 1997).
In the present study, we have studied the CL, LCN, and CL-LCN mouse strains in an assessment of the relative roles of liver and small intestine in the first-pass metabolism of nifedipine as a model drug. Nifedipine is a calcium-channel antagonist used for the treatment of hypertension and other cardiovascular disorders (Echizen and Eichelbaum, 1986). We tested the hypothesis that small intestinal P450 enzymes play an important role in regulating the bioavailability of oral nifedipine. Initial studies were performed on CL mice. Small intestinal microsomes from control and CL mice were examined to determine the extent of decreases in Cpr expression and in the rates of microsomal metabolism of nifedipine to oxidized nifedipine (NFPO). Intestinal expression of selected P450 proteins was also examined to detect any compensatory increases in the CL mice. Pharmacokinetic parameters of nifedipine clearance were determined after oral or intraperitoneal treatment in each of the mouse models. Our findings indicated that the decreased Cpr expression in the CL mice led to increases, relative to the control B6 mice, in systemic bioavailability of both oral and intraperitoneal nifedipine. Furthermore, the additional decrease of extrahepatic Cpr expression in the CL-LCN mice, compared with that in the LCN mice, led to further increases in systemic bioavailability of oral and intraperitoneal nifedipine. Importantly, in both studies, the magnitudes of the increase in systemic bioavailability were greater for oral than for intraperitoneal nifedipine, a finding that strongly supports the role of small intestinal P450 enzymes in the first-pass clearance of oral nifedipine.
Materials and Methods
Animals. C57BL/6 (B6) mice, which were used as the wild-type control strain for the CL mice, were obtained from breeding stocks maintained at the Wadsworth Center. LCN (B6/N10) and WT control littermates (Cprlox/lox), CL (B6/N1), and CL-LCN (B6/N1.5) mouse models have been described recently (Gu et al., 2003, 2007; Wu et al., 2003, 2005). All studies with mice were approved by the Wadsworth Center Institutional Animal Care and Use Committee.
Isolation of Intestinal Epithelial Cells and Preparation of Microsomes. Tissues from two to three mice of a given strain were combined for each microsomal preparation. Intestinal epithelial cells (enterocytes) were isolated, and microsomes were prepared as reported previously (Zhang et al., 2003). Liver microsomes were prepared essentially according to Fasco et al. (1993) but with use of protease inhibitors (Sigma-Aldrich, St. Louis, MO), as described for the preparation of small intestinal microsomes (Zhang et al., 2003). Microsomes were stored at -80°C until use. Microsomal protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce Chemical, Rockford, IL) with bovine serum albumin as the standard.
Immunoblot Analysis. For immunoblot analysis, microsomal proteins were separated on NuPAGE Bis-Tris gels (10%) (Invitrogen, Carlsbad, CA). Polyclonal antibodies to rat CYP1A1/2, CYP2B1, CYP2C6, or CYP3A2 were purchased from BD Bioscience (Bedford, MA). Polyclonal rabbit anti-rat CPR antibody was obtained from BD Gentest (Woburn, MA), Peroxidase-labeled rabbit anti-goat IgG or goat anti-rabbit IgG (Sigma-Aldrich) was detected with an enhanced chemiluminescence kit (Amersham, Arlington Heights, IL). The optical densities of detected bands were determined using a Personal Densitometer SI (Molecular Dynamics, Sunnyvale, CA).
In Vitro Metabolism of Nifedipine. The nifedipine oxidase assay was performed essentially as described (Stresser et al., 2000) with some modifications. Nifedipine (Sigma-Aldrich) was incubated for various periods of time at 37°C in a 500-μl reaction mixture containing 25 mM potassium phosphate buffer, pH 7.4, 1.0 mM NADPH, and 0.25 mg of microsomal protein. The P450 contents were 0.19 ± 0.05, 0.25 ± 0.03, 1.0 ± 0.1, and 1.6 ± 0.2 nmol/mg protein (means ± S.D., n = 3), for intestinal microsomes from B6 and CL mice and liver microsomes from B6 and CL mice, respectively. The reaction was initiated by the addition of NADPH, and it was terminated by the addition of 50 μl of 1.0 M NaOH to the reaction mixture. Control experiments were performed in which NADPH was omitted. Metabolites were extracted according to the method of Jankowski and Lamparczyk (1994), with 2.5 ml of hexane-dichloromethane (70:30, v/v). Nitrendipine (0.5 nmol; Sigma-Aldrich) was added as the internal standard for monitoring extraction efficiency. After centrifugation at 1500g for 10 min, the organic layer was transferred to a new tube and was evaporated under nitrogen. The residue was reconstituted with 200 μl of 70% methanol and 30% water containing 0.085% phosphoric acid, and 100-μl aliquots were taken for HPLC analysis.
For HPLC analysis of nifedipine and the nifedipine metabolite NFPO (Ultrafine Chemicals, Manchester, UK), a Waters Nova-Pak C18 column (8 × 100 mm), preceded by a C18 precolumn cartridge, was used in all assays. A Waters 2690 Alliance HPLC system and a Waters 996 model photodiode array detector were used. The solvent system comprised solvent A (0.085% phosphoric acid) and solvent B (100% methanol). A 6-min linear gradient from 45% B to 62% B was applied at a flow rate of 1 ml/min, followed by a 14-min isocratic elution at 62% B and then a 5-min wash at 100% B, before return to the starting condition. NFPO, identified on the basis of comigration with authentic standards, was detected by monitoring of UV absorbance at 230 nm, and it was quantified by comparison of peak areas to a standard curve. The retention times for NFPO, nifedipine, and nitrendipine were ∼8.4, 10, and 14 min, respectively. Enzyme kinetics were examined with nifedipine concentrations at 5 to 50 μM.
Pharmacokinetics Analysis. Mice (five to six in each group) were treated with nifedipine, either intraperitoneally (5 or 10 mg/kg) or orally through gavage (10 or 20 mg/kg). Blood samples were collected from the tail at various times after nifedipine administration, as described recently (Gu et al., 2005). Nifedipine was extracted from whole blood, using conditions described by Jankowski and Lamparczyk (1994). The nifedipine concentration was determined by HPLC as described above. Pharmacokinetic parameters were calculated using WinNonlin software (version 5.0.1; Pharsight, Mountain View, CA). Statistical significance of differences between groups was examined using Student's t test. For multiple group comparisons, one-way analysis of variance was performed, followed by Tukey's test (for pairwise comparisons), or for samples failing a normality test or a variance test, the Kruskal-Wallis analysis of variance on ranks was performed, followed by Dunn's test (for pairwise comparisons) (SigmaStat software; SPSS Inc., Chicago, IL), as indicated in the figure legends or the notes to the tables.
Results
CPR and P450 Expression in the Small Intestine of CL Mice. The expression of CPR in mouse small intestine was examined by immunoblot analysis with an antibody to rat CPR. As shown in Fig. 1A, the levels of CPR protein in small intestinal microsomes from the CL mice were greatly reduced relative to those from B6 mice. Quantitative immunoblot analysis of CPR protein levels indicated that the extent of decrease in the CL mice, relative to that in the B6 mice, was ∼90% at the age of 3 months. Decreases in CPR expression were also observed in 8-month-old CL mice (data not shown).
As shown in Fig. 1, B through E, the reduced expression of CPR protein occurred concomitantly with increased expression of several P450 proteins in the small intestine, as we had previously found in the liver (Weng et al., 2005; Wu et al., 2005), of the CL mice. A comparison of the levels of small intestinal P450 expression between the CL and B6 mice indicated that the extents of increase in P450 expression were ∼4-fold (CYP1A1), 2- to 3-fold (CYP2B), 3- to 4-fold (CYP2C), and 1.5-fold (CYP3A). Because the antibodies to CYP2B1, CYP2C6, and CYP3A2 cross-react with more than one member of the same CYP subfamily and possibly with members of other subfamilies, the signals detected represent the sum of all recognized forms. For CYP1A, although the antibodies to rat CYP1A react with both mouse CYP1A1 and CYP1A2, the two isoforms were resolved on gels (data not shown), thus permitting specific detection of CYP1A1.

Immunoblot analysis of CPR and P450 proteins expressed in the small intestine of B6 and CL mice. For each mouse strain, enterocytes from three male, 3-month-old mice were pooled for microsomal preparation. Microsomal proteins (10 μg each), loaded in duplicate, were analyzed on immunoblots with antibodies to CPR, CYP1A1/2, CYP2B, CYP2C, or CYP3A.
In Vitro Metabolism of Nifedipine by Hepatic and Intestinal Microsomes of CL Mice. The impact of the reduced CPR expression on microsomal P450 activities toward nifedipine was examined in the CL mice and wild-type (B6) controls with nifedipine at 5 to 50 μM. Despite the compensatory increases in the expression of P450 isoforms potentially involved in nifedipine metabolism (e.g., CYP3A), the maximal rates of formation of the nifedipine metabolite NFPO in liver and small intestinal microsomes were lower in the CL mice than in the B6 mice (Table 1). The extent of decrease in microsomal metabolic activity was similar to reductions found previously in the hepatic microsomal metabolism of acetaminophen and testosterone in CL mice (Wu et al., 2005). For liver microsomes, the reduced CPR expression lowered Vmax values for NFPO formation from 0.84 nmol/min/mg in B6 to 0.48 nmol/min/mg in CL mice, but it did not decrease catalytic efficiency (Vmax/Km), given that the apparent Km values were also lowered (from 17.4 to 7.1 μM). For the small intestinal microsomes, the results were similar to those in the liver: the apparent Km and Vmax values were 18.3 μM and 0.70 nmol/min/mg, respectively, for samples prepared from B6 mice and 11.2 μM and 0.33 nmol/min/mg, respectively, for samples prepared from the CL mice. The catalytic efficiency was essentially the same for the two mouse strains.
Kinetic parameters for nifedipine metabolism catalyzed by hepatic and small intestinal microsomes from B6 and CL mice
Rates of formation of NFPO were determined as described under Materials and Methods. Reaction mixtures contained 25 mM phosphate buffer, pH 7.4, 5 to 50 μM nifedipine, and 0.5 mg/ml microsomal protein. Each microsome preparation was obtained from pooled tissues from two to three mice. Reactions were performed in duplicate in the presence or absence of 1.0 mM NADPH. Rates of product formation were linear with reaction time. The values presented for the apparent Km and apparent Vmax are the means of two independent determinations (with the individual values listed in parentheses), except for CL liver, for which a single microsomal preparation was analyzed.
In Vivo Metabolism of Nifedipine in CL and B6 Mice. To investigate the impact of the reduced CPR expression and the consequent reduction in microsomal nifedipine oxidase activity on nifedipine bioavailability, we performed a pharmacokinetics study on B6 and CL mice. The concentration-time curves for plasma nifedipine after a single-dose administration of nifedipine are shown in Fig. 2. The pharmacokinetic parameters are shown in Table 2. After oral administration of nifedipine at 20 mg/kg, the absorption of nifedipine was rapid in both B6 and CL mice. However, the t½ and AUC values were 7.0- and 4.0-fold, respectively, higher in the CL mice than in the B6 mice, indicating that nifedipine disposition was much slower in the CL mice than in the B6 mice, when nifedipine was administered orally. After a single intraperitoneal administration of nifedipine at 10 mg/kg, the increases in the t½ and AUC values were not as remarkable as those for oral nifedipine, being only 1.2- and 1.8-fold, respectively, and higher in the CL mice than in the B6 mice. Taken together, the differing extents of decreases in nifedipine clearance in the CL mice, between oral and intraperitoneal routes of administration, suggested that small intestinal P450-catalyzed nifedipine metabolism plays an important role in controlling systemic bioavailability of oral nifedipine.
Pharmacokinetic parameters for nifedipine clearance
Blood nifedipine levels (from Fig. 2) were used to calculate pharmacokinetic parameters, including AUC, Tmax, Cmax, and t½. Values presented are means ± S.D. (n = 5-6 for each strain). Student's t test was used for statistical analysis of differences between B6 and CL strains.
In Vivo Metabolism of Nifedipine in LCN and CL-LCN Mice. The fact that hepatic metabolism of nifedipine was decreased concurrently with decreases in small intestinal nifedipine metabolism in the CL mouse model could confound the deduced role of the small intestine in controlling systemic bioavailability of oral nifedipine. CPR expression in other extrahepatic tissues is also reduced; however, the contributions of these tissues to systemic clearance of either oral or intraperitoneal nifedipine are expected to be less important than is the hepatic contribution. To more clearly demonstrate the influence of intestinal P450-catalyzed nifedipine metabolism on systemic bioavailability of oral nifedipine, we further studied the clearance of oral and intraperitoneal nifedipine in mouse models that are deficient in Cpr expression in hepatocytes. Accordingly, pharmacokinetic studies were performed with LCN and CL-LCN mice (and wild-type controls) after oral or intraperitoneal administration of nifedipine. Although both LCN and CL-LCN mice have liver-specific Cpr deletion, the small intestinal nifedipine metabolism in LCN mice should be the same as that in wild-type mice, whereas in CL-LCN mice, the small intestinal nifedipine metabolism is decreased to the same extent as that in the CL mice. In this experiment, the nifedipine dose used (10 mg/kg for oral and 5 mg/kg for intraperitoneal) was lower than the doses that had been used for the CL mice, because higher doses were not tolerated by the LCN and CL-LCN mice in pilot studies. As shown in Table 3 and Fig. 3, in the LCN mice, the time to the peak blood nifedipine concentration (Tmax) after a single oral dose of nifedipine (10 mg/kg) was delayed by ∼3 h, and the maximum concentration (Cmax), the elimination half-life (t½), and AUC values were 2.2, 3.7, and 6.9 times, respectively, higher than those in WT mice. These data demonstrated that the hepatic microsomal P450-catalyzed nifedipine metabolism plays a primary role in controlling nifedipine clearance in wild-type mice. However, in CL-LCN mice, Tmax was further delayed to ∼6 h, and the Cmax and AUC values were 80 and 120%, respectively, higher than those in the LCN mice. These results confirmed in mouse models with little hepatic P450 activity that a decrease in the level of CPR protein in the small intestine and other extrahepatic tissues led to further increases in the bioavailability of oral nifedipine. Because the small intestine is the site of absorption for oral drugs and because it often has much higher overall P450 metabolic activity than do the other extrahepatic organs, these data provide strong support for the hypothesis that small intestinal P450-catalyzed nifedipine metabolism plays an important role in controlling the bioavailability of oral nifedipine. In contrast, after a single intraperitoneal dose of nifedipine (5 mg/kg), the time to the peak blood nifedipine concentration and the Cmax were increased to similar extents in the LCN and CL-LCN mice compared with the WT mice. The t½ and AUC values in LCN mice were 9.7- and 16.8-fold, respectively, higher than those in WT mice, confirming the critical role of hepatic microsomal P450-catalyzed nifedipine metabolism in systemic clearance of intraperitoneal nifedipine. In CL-LCN mice, the t½ and AUC values were further increased, when compared with the corresponding values for the LCN mice; however, the extent of increase in the AUC value (60%) was lower for intraperitoneal than for oral (120%) nifedipine, a finding consistent with a greater role of the small intestine for first-pass clearance of oral drugs than for clearance of systemic drugs. These results also suggest that the small intestinal P450-catalyzed nifedipine metabolism can contribute to systemic clearance of intraperitoneally administered nifedipine, probably through enterohepatic circulation.
Pharmacokinetic parameters for nifedipine clearance in WT, LCN, and CL-LCN mice
Blood nifedipine levels (from Fig. 3) were used to calculate pharmacokinetic parameters, including AUC, Tmax, Cmax, and t½. Values presented are means ± S.D. (n = 5-6 for each strain). Student's t test was used for statistical analysis of differences between the WT and LCN or between LCN and CL-LCN strains.

Plasma concentrations of nifedipine as a function of time, after a single dose of nifedipine in B6 and CL mice. Two- to 4-month-old B6 and CL mice (male or female) were given a single oral (20 mg/kg) (top) or intraperitoneal (10 mg/kg) (bottom) dose of nifedipine in 20% dimethyl sulfoxide and 80% corn oil. Blood samples were collected from individual mice at 0 to 24 h after injection. Nifedipine was determined by HPLC, as described under Materials and Methods. Values represent the means ± S.D. (n = 5-6). Student's t test was used for statistical analysis. *, CL is significantly different from B6 (p < 0.01); **, CL is significantly different from B6 (p ≤ 0.05).
Discussion
Oral administration of xenobiotics initially leads to small intestinal metabolism, whereas intraperitoneal administration initially leads to hepatic metabolism. A comparison of the data from oral administration and intraperitoneal injection provides insight into the role of small intestinal metabolism in controlling systemic bioavailability of various drugs. The observed difference in nifedipine bioavailability between CL and B6 mice (4- and 2-fold increases in AUC for oral and intraperitoneal nifedipine, respectively) reflects differing P450 activities in liver as well as in extrahepatic tissues in the two mouse strains, whereas the observed difference in nifedipine bioavailability between CL-LCN and LCN mice (120 and 60% increases in AUC for oral and intraperitoneal nifedipine, respectively) reflects contributions of P450 enzymes expressed in all extrahepatic tissues, including the small intestine. For orally administered drugs, the extrahepatic contributions in these two comparisons represent mainly those of the small intestine. P450 enzymes in enterocytes, the site of initial absorption for oral drugs and thus a site where the drug concentration will be higher than that in other extrahepatic tissues, are more likely to play a substantial role in the disposition of an oral drug than are enzymes in other extrahepatic tissues. Therefore, the greater magnitudes of increase in systemic bioavailability for oral nifedipine than for intraperitoneal nifedipine observed in these two pharmacokinetics experiments support the proposed role of small intestinal P450 enzymes in the first-pass clearance of oral nifedipine.
It has long been hypothesized that the small intestine plays a significant role in the first-pass extraction of oral nifedipine (Bailey et al., 1991). Supporting evidence for this idea came primarily from pharmacokinetic studies of nifedipine administered via various routes and from studies of in vivo drug-drug interactions. The major relevant findings include 1) alteration of bioavailability of oral nifedipine by presystemic extraction in small intestine in pharmacokinetic studies in rats (Grundy et al., 1997), 2) increased bioavailability of oral nifedipine in rats suffering from decreased intestinal P450 and P450 reductase contents as a result of repeated exposure to the anticancer drug 5-fluorouracil (Yoshisue et al., 2001), 3) increased bioavailability of oral nifedipine in humans (Bailey et al., 1991, 1998) and rats (Grundy et al., 1997, 1998; Mohri et al., 2000) given concentrated grapefruit juice (or active components), which inhibits intestinal, but apparently not hepatic, CYP3A-mediated drug metabolism, and 4) a reduction in oral, but not intravenous, nifedipine bioavailability in patients during coadministration with rifampin, a CYP3A inducer (Holtbecker et al., 1996). Nonetheless, the effects of the interacting drugs in these studies were not strictly specific to P450 enzymes or to the intestine. For example, grapefruit juice components can have inhibitory effects on drug absorption (Grundy et al., 1998; Odou et al., 2005) or efflux transport (Edwards et al., 1999; Honda et al., 2004), in addition to inhibitory effects on intestinal CYP3A activity (Edwards et al., 1999). In this regard, nifedipine is a likely substrate for P-glycoprotein, based on the observed ability of nifedipine to inhibit drug transport in Caco-2 cells (Kim et al., 1999), although nifedipine failed to inhibit drug transport in porcine kidney LLC-PK1 cells that overexpressed P-glycoprotein (Katoh et al., 2001). Therefore, although the ability of the small intestine to metabolize and extract absorbed nifedipine has been demonstrated in a rat model of in situ intestinal single-pass perfusion (Iwao et al., 2002), more definitive in vivo studies, in which the P450 system is specifically modulated in a tissue-selective fashion, are needed, to enable a quantitative assessment of the role of small intestinal P450-mediated oxidation in nifedipine bioavailability.
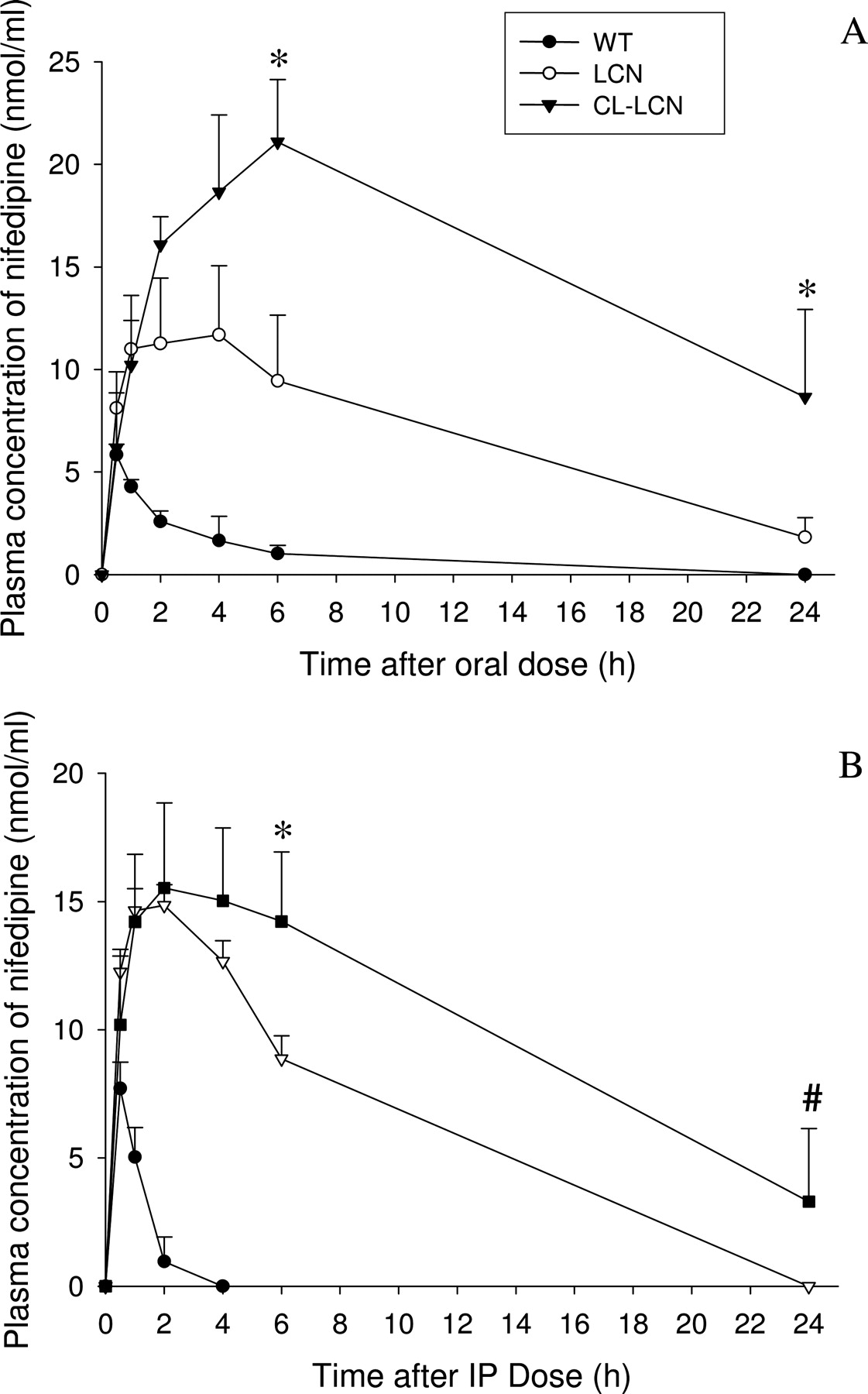
Plasma concentrations of nifedipine as a function of time, after a single dose of nifedipine in WT, LCN, and CL-LCN mice. Two- to 4-month-old, WT, LCN, and CL-LCN mice (male or female) were given a single oral (10 mg/kg) (A) or intraperitoneal (5 mg/kg) (B) dose of nifedipine in 20% dimethyl sulfoxide and 80% corn oil. Blood samples were collected from individual mouse at 0 to 24 h after injection. Values represent the means ± S.D. (n = 5-6). *, CL-LCN is significantly different from LCN (p < 0.01; Tukey's test). #, CL-LCN is significantly different from LCN (p < 0.05; Dunn's test). The values for LCN and CL-LCN groups were significantly greater than those for WT for all except the first time points after dosing.
In our study, we have, for the first time, clearly demonstrated the in vivo contribution of CPR/P450-mediated metabolism in the clearance of nifedipine. Although several non-P450 enzymes, such as heme oxygenases, in addition to microsomal P450 enzymes, depend on CPR for function (Gu et al., 2003), none of these enzymes is directly involved in drug metabolism. Therefore, the impact of reduced CPR expression on microsomal drug metabolism can be unambiguously attributed to the associated decreases in microsomal P450 activity. In this regard, we have found, through a microarray analysis of gene expression in the small intestine, that none of the multidrug resistance genes, including that encoding the P-glycoprotein, is significantly changed in the CL mice, compared with the wild-type mice (Y. Weng and X. Ding, unpublished data).
The extent of decrease in CPR protein levels in the small intestine of the CL mice, which had not been examined previously, was found to be ∼90% (compared with that of B6 mice), an extent similar to the decreases in the brain and lung, but much greater than the decreases in the liver (∼70%) (Wu et al., 2005). As was found previously in the liver and kidney (Weng et al., 2005; Wu et al., 2005), the reduction in CPR expression in the CL mice led to significant increases in P450 expression in the small intestine. Among the P450 forms examined, CYP1A1 was increased 4-fold, CYP2B and CYP2C were increased 2- to 4-fold, and CYP3A was increased 1.5-fold. In previous studies, levels of CYP1A2, but not of CYP1A1, were found to be increased in the livers of LCN and CL mice, relative to those of control mice (Gu et al., 2003; Weng et al., 2005). In contrast, we found in the present study that CYP1A1 is induced in the small intestine, a site in which, as we have demonstrated earlier (Zhang et al., 2003), CYP1A2 is not expressed. This apparently tissue-selective induction of CYP1A1 in the small intestine can perhaps be explained by a greater accumulation in the small intestine of dietary aryl hydrocarbon receptor ligands that are normally degraded by P450 enzymes or by a greater sensitivity of intestinal CYP1A1 expression to induction through the aryl hydrocarbon receptor. Further mechanistic studies to differentiate these possibilities are warranted.
The major in vivo metabolite of nifedipine is NFPO, which is produced by CYP3A4, and, to a lesser extent, by CYP1A2 and 2A6 (Guengerich et al., 1986). In the CL mice, the rates of in vitro microsomal metabolism of nifedipine were significantly decreased, relative to those in B6 mice, in both liver and small intestine, a finding contrasting with the recently reported lack of a decrease in cyclophosphamide 4-hydroxylase activities in liver microsomes from the CL mice (Gu et al., 2007). This difference in the impact of Cpr-low status on hepatic microsomal P450 activity toward nifedipine and cyclophosphamide most likely reflects the differing extents of the compensatory increases in expression of those P450 isoforms that are principally involved in the metabolism of these two drugs. In this regard, although previous studies have shown that cyclophosphamide 4-hydroxylation is catalyzed mainly by CYP2B (Clarke and Waxman, 1989), whereas nifedipine is metabolized mainly by CYP3A (Guengerich et al., 1986), we note that the primary mouse CYP isoforms involved in these reactions in the liver have not been identified. In the small intestine, the substantial induction of CYP1A1 in the CL mice may make it impractical to study the role of intestinal P450s in the metabolism of drugs or other xenobiotic compounds that are mainly CYP1A1 substrates in this model. On the other hand, the relatively low induction of CYP3A, coupled with the low residual CPR expression in the small intestine, will probably result in significant decreases in the rates of microsomal metabolism of drugs that are CYP3A substrates, as was observed here for nifedipine oxidation. Thus, it should be possible to use the combination of the LCN and CL-LCN mouse models to test the roles of small intestinal P450 enzymes in the first-pass metabolism of other oral drugs, particularly those metabolized by CYP3A. Nonetheless, because of the relatively high residual P450 activities in the intestinal microsomes of the CL and CL-LCN mice, the extents determined in these models for the intestinal contribution to the first-pass drug clearance will probably be underestimates. Studies to develop an intestinal epithelium-specific Cpr-null mouse model are in progress.
In the current study, doses of 10 to 20 mg/kg were used to examine the contribution of small intestinal P450 enzymes to the first-pass clearance of oral nifedipine. The therapeutic dose of oral nifedipine is <1 mg/kg in human patients (Holtbecker et al., 1996). It is conceivable that, at lower doses (doses close to the therapeutic level), the small intestinal P450 systems will have a greater impact on systemic bioavailability of oral nifedipine than was found in the present study. When nifedipine is given at a high dose, the intestinal P450 system is more likely to be saturated, thus allowing greater amounts of drugs entering into the circulation.
Acknowledgments
We gratefully acknowledge the use of the services of the Biochemistry and Molecular Genetics Core Facilities of the Wadsworth Center. We thank Dr. Adriana Verschoor for reading the manuscript, and Weizhu Yang for assistance with mouse breeding.
Footnotes
-
This work was supported in part by U.S. Public Health Service Grants CA092596 and ES07462 from the National Institutes of Health.
-
doi:10.1124/dmd.107.016543.
-
ABBREVIATIONS: P450, cytochrome P450; CPR, NADPH-cytochrome P450 reductase; CL, Cpr-low; LCN, liver-Cpr-null; CL-LCN, Cpr-low and liver-Cpr-null; NFPO, oxidized nifedipine; WT, wild type; HPLC, high performance liquid chromatography; AUC, area under the curve.
- Received May 8, 2007.
- Accepted June 11, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}