


Abstract
Nevirapine, a non-nucleoside HIV-1 reverse transcriptase inhibitor, has been associated with incidences of skin rash and hepatotoxicity in patients. Although the mechanism of idiosyncratic hepatotoxicity remains unknown, it is proposed that metabolic activation of nevirapine and subsequent covalently binding of reactive metabolites to cellular proteins play a causative role. Studies were initiated to determine whether nevirapine undergoes cytochrome P450 (P450)-mediated bioactivation in human liver microsomes to electrophilic intermediates. Liquid chromatography-tandem mass spectrometry analysis of incubations containing nevirapine and NADPH-supplemented microsomes in the presence of glutathione (GSH) revealed the formation of a GSH conjugate derived from the addition of the sulfydryl nucleophile to nevirapine. No other GSH conjugates were detected, including conjugates of oxidized metabolites of nevirapine. These findings are consistent with a bioactivation sequence involving initial P450-catalyzed dehydrogenation of the aromatic nucleus with a 4-methyl group in nevirapine to an electrophilic quinone methide intermediate, which is subsequently attacked by glutathione yielding the sulfydryl conjugate. Formation of the nevirapine GSH conjugate was primarily catalyzed by heterologously expressed recombinant CYP3A4 and, to a lesser extent, CYP2D6, CYP2C19, and CYP2A6. In addition, the quinone methide reactive metabolite was a mechanism-based inactivator of CYP3A4, with inactivation parameters KI = 31 μM and kinact = 0.029 min–1, respectively. It is proposed that formation of the quinone methide intermediate may represent a rate-limiting step in the initiation of nevirapine-mediated hepatotoxicity.
Nevirapine (NVP) (Fig. 1) is the first non-nucleoside reverse transcriptase inhibitor approved by the U.S. Food and Drug Administration (FDA) for use in combination therapy of HIV-1 infection. Currently, nevirapine remains one of the most prescribed antiretroviral drugs in the developing countries, both to prevent vertical transmission and in combination therapy (Lockman et al., 2007). Despite its therapeutic benefits, treatment with nevirapine has been associated with a significant incidence of hepatotoxicity and skin rash (Pollard et al., 1998). An FDA black box warning label was placed on nevirapine in 2000 due to concerns regarding its hepatotoxicity. Although the exact mechanism of nevirapine hepatotoxicity is not clearly understood, a probable causal link between nevirapine metabolites and skin rash has been established (Popovic et al., 2006).
In humans, nevirapine undergoes significant hepatic metabolism mainly by 2-, 3-, 8-, and 12-hydroxylation followed by glucuronidation of these hydroxyl metabolites (Riska et al., 1999). The 2-, 3-, and 8-hydroxylation can presumably lead to formation of para-quinone imine intermediates after further oxidation (Fig. 1). The metabolite 12-hydroxynevirapine has the potential to be sulfated followed by loss of sulfate to form a reactive quinone methide (Fig. 1). In addition, 4-carboxynevirapine, formed from further oxidation of 12-hydroxynevirapine, can generate acyl glucuronide, which has the potential to bind to cellular proteins. In human liver microsomal incubations, similar hydroxylation metabolites were observed (Erickson et al., 1999). CYP3A4 was identified as the major enzyme involved in formation of 2- and 12-hydroxynevirapine, whereas CYP2B6 was the predominant enzyme forming 3- and 8-hydroxynevirapine (Erickson et al., 1999). However, no glutathione (GSH) conjugates of nevirapine or its hydroxyl metabolites have been reported, and the identities of putative reactive metabolites of nevirapine remain unknown.
More recently, 12-hydroxynevirapine has been proposed as a factor in nevirapine hepatocarcinogenicity (Antunes et al., 2008) as well as skin rash (Chen et al., 2008). The reactivity of 12-sulfoxynevirapine was demonstrated by formation of multiple DNA adducts in the reactions with 12-mesyloxynevirapine (Antunes et al., 2008). The observation that 12-hydroxynevirapine caused skin rash at a lower dose than nevirapine suggested that this metabolite is responsible for the rash (Chen et al., 2008). Meanwhile, lower incidence of rash was demonstrated with 12-trideutero-nevirapine (DNVP), which decreased the rate of 12-hydroxylation due to deuterium isotope effects (Chen et al., 2008). In this study, we report a GSH conjugate derived from the addition of the sulfydryl nucleophile to nevirapine. This adduct was primarily formed by CYP3A4, which was inactivated in a time- and concentration-dependent manner by nevirapine. These data are important for further understanding the relationship between metabolic activation and hepatotoxicity of nevirapine.

Proposed bioactivation pathways of the non-nucleoside reverse transcriptase inhibitor nevirapine.
Materials and Methods
Materials. The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO): GSH, NADPH, potassium tert-butoxide, DMSO-d6, midazolam, and 1′-hydroxymidazolam. NVP was obtained from US Pharmacopeia (Rockville, MD). Pooled human liver microsomes (HLMs) and Supersomes containing cDNA-baculovirus-insect cell-expressed cytochromes P450 (P450s; CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) were obtained from BD Gentest (Woburn, MA). Formic acid, methanol, and acetonitrile were purchased from EM Scientific (Gibbstown, NJ).
Synthesis of DNVP. Synthesis of DNVP was carried out according to the procedure described previously (Chen et al., 2008). In brief, NVP (0.2 g, 0.75 mmol) and potassium tert-butoxide (0.17 g, 1.5 mmol) were dissolved in DMSO-d6 (5 ml, 71.41 mmol) in a flame-dried flask. The reaction mixture was refluxed at 140°C under argon for 48 h before it was diluted with water (20 ml) and extracted with ethyl acetate (20 ml × 2). The ethyl acetate layer was then washed with brine, dried over anhydrous Na2SO4, and concentrated to yield crude product. The crude product was column purified using ethyl acetate to yield 0.16 g of a yellow solid product. 1H-NMR (CDCl3): 0.29 to 0.40 ppm (2H, m), 0.81 to 0.90 ppm (2H, m), 3.57 to 3.61 ppm (1H, m), 7.01 ppm (1H, d, J = 4.8 Hz), 7.12 ppm (1H, dd, J = 4.8 Hz, J = 7.6 Hz), 7.97 ppm (1H, dd, J = 2.2 Hz, J = 6.8 Hz), 8.03 ppm (1H, d, J = 4.8 Hz), 8.41 ppm (1H, dd, J = 2.0 Hz, J = 4.8 Hz) and 9.84 ppm (1H, bs). High-resolution mass spectra (m/z) was determined as 270.1431 (M + H). Based on the ratio of peak heights in mass spectrometry analysis, the isotopic purity was estimated as ∼83% and synthesized DNVP contained only traces of NVP.
Instrumentation. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses were performed on an API 4000 Q-Trap mass spectrometer (Applied Biosystems, Foster City, CA) as described previously (Wen et al., 2008), with minor modifications. In brief, the precursor ion (PI) scan of m/z 272 was run in the negative mode with 0.2 Da step size, 5 ms pause between mass ranges, and 2 s scan rate or 50 ms dwell. The enhanced production (EPI) scans were run in the positive mode at a scan range for daughter ions from m/z 100 to 1000, followed by the MS3 scans in the positive mode at a scan range from m/z 100 to 350. Negative EPI scans were carried out in a separate run at a scan range from m/z 100 to 1000. Data were processed using Analyst 4.1 software (Applied Biosystems). A Shimadzu high-performance liquid chromatography (HPLC) system (Shimadzu Scientific Instruments, Columbia, MD) was coupled with an Agilent Eclipse XDB-Phenyl C18 column (3.0 × 150 mm, 3.5 μm; Agilent Technologies, Santa Clara, CA). The HPLC mobile phase A was 5 mM ammonium acetate in water with 0.1% formic acid, and mobile phase B was acetonitrile with 0.1% formic acid. A Shimadzu LC-20AD solvent delivery module (Shimadzu Scientific Instruments) was used to produce the following gradient elution profile: 5% solvent B for 2 min, followed by 5 to 70% B in 20 min, and 70 to 90% B in 2 min. The HPLC flow rate was 0.3 ml/min. For relative comparison of GSH adduct levels formed by individual recombinant P450 enzymes, the mass spectrometer was operated in the multiple reaction monitoring mode. Multiple reaction monitoring transitions were simultaneously monitored as follows: for the NVP GSH adduct, m/z 572→443 and 572→299; and for the DNVP GSH adduct, m/z 574→445 and 574→301. NMR analysis was recorded on a Bruker AV-300 NMR (Bruker AXS Inc., Madison, WI), and high-resolution mass spectrometry was performed on a LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA).

LC-MS/MS analysis of the NVP GSH adduct. A, negative precursor ion scanning at m/z 272; B, MS/MS spectrum at m/z 572 in positive ion mode; C, MS3 mass spectrum of the fragment ion at m/z 299 in positive ion mode.
Microsomal Incubations. All incubations were performed at 37°C in a water bath. Stock solutions of the test compounds were prepared in methanol. The final concentration of methanol in the incubation was 0.1% (v/v). Pooled HLMs and the human cDNA-expressed P450 isozymes were carefully thawed on ice before the experiment. NVP or DNVP (10 μM) was mixed with 1 mg/ml HLM proteins in 100 mM potassium phosphate buffer (pH 7.4) supplemented with 1 mM GSH. The total incubation volume was 1 ml. After 3-min preincubation at 37°C, the incubation reactions were initiated by the addition of 1 mM NADPH. Reactions were terminated by the addition of 150 μl of trichloroacetic acid (10%) after 60-min incubation. Incubations with the recombinant cDNA-expressed P450 isozymes were performed similarly except that liver microsomes were substituted by BD Gentest Supersomes (100 pmol/ml). Control samples containing no NADPH or substrates were included. Samples were centrifuged at 10,000 g for 15 min at 4°C to pellet the precipitated proteins, and supernatants were subjected to LC-MS/MS analysis of GSH adducts. Deuteriums of DNVP were not exchangeable under these conditions, as indicated by mass spectrometric analysis. Each incubation was performed in triplicate. For the negative precursor ion scanning of GSH adducts, supernatants were concentrated by solid phase extraction as described previously (Wen et al., 2008), before LC-MS/MS analyses.
Enzyme Inactivation. Midazolam 1′-hydroxylase activity was determined to quantify time- and concentration-dependent loss of CYP3A4 activity of HLMs in the presence of nevirapine. Primary incubations included NVP (0, 1, 3, 10, 30, 50, and 100 μM), 1 mM NADPH, 1 mg/ml HLM, 3 mM MgCl2, and 0.1 M potassium phosphate buffer (pH 7.4). The mixture was incubated in a 37°C shaking water bath for various time points (0, 2, 6, 12, and 20 min). At each preincubation time point, 10-μl aliquots of the primary incubation mixtures were transferred to a secondary incubation to a final volume of 100 μl, which included 10 μM midazolam, 1 mM NADPH, 3 mM MgCl2, and 0.1 M potassium phosphate (pH 7.4). The midazolam mixture was incubated in a 37°C shaking water bath for 5 min and stopped by the addition of ice-cold acetonitrile in a 1:1 ratio (v/v). The mixture was centrifuged at 10,000 g for 15 min. The supernatant (10 μl) was injected and analyzed by a Sciex API 4000 triple quadrupole mass spectrometer (Applied Biosystems) coupled with a Shimadzu HPLC system (Shimadzu Scientific Instruments). 1′-Hydroxymidazolam was separated using a Hypersil BDS C18 column (50 × 2.1 mm, 5 μm; Thermo Fisher Scientific) at a flow rate of 0.3 ml/min. Selective reaction monitoring experiments in the positive ionization mode were performed using a dwell time of 150 ms per transition to detect the following precursor (Q1) to product (Q3) ion pairs: 326 to 291 and 342 to 203 for midazolam and 1′-hydroxymidazolam, respectively. The HPLC mobile phase A was 5 mM ammonium acetate in water with 0.1% formic acid, and mobile phase B was acetonitrile with 0.1% formic acid. A Shimadzu LC10AD solvent delivery module (Shimadzu Scientific Instruments) was used to produce the following gradient elution profile: 10% solvent B for 0.5 min, 10 to 90% B in 3.5 min, followed by 90% B for 1 min.

LC-MS/MS analysis of the DNVP GSH adduct. A, negative precursor ion scanning at m/z 272; B, MS/MS spectrum at m/z 574 in positive ion mode; C, MS3 mass spectrum of the fragment ion at m/z 301 in positive ion mode.
Results
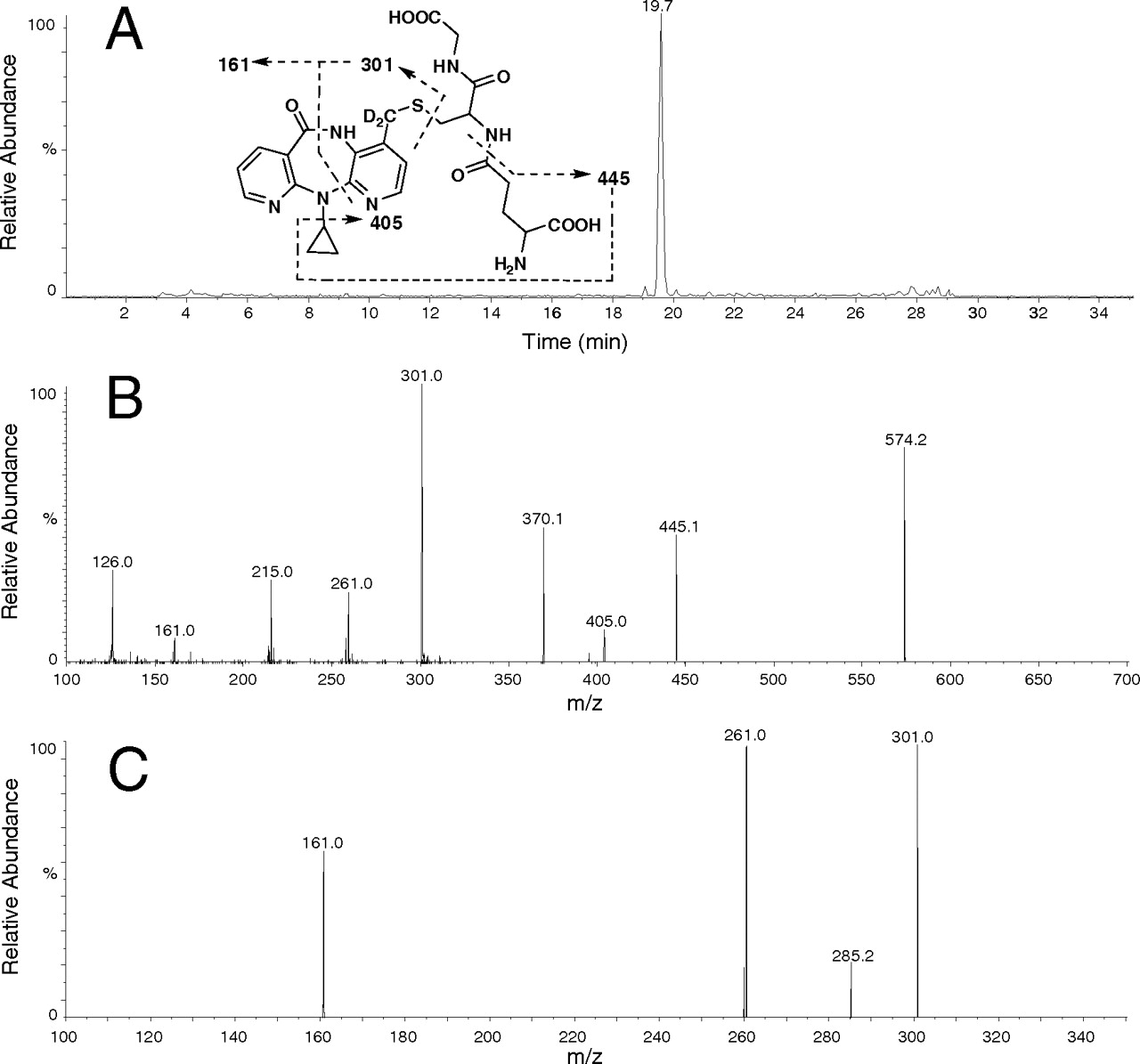
Characterization of the Nevirapine GSH Adduct. As shown in Fig. 2A, only one component was detected by the negative precursor ion scanning of m/z 272. This peak was eluted at 19.7 min and was not detected when either nevirapine or NADPH was absent from the incubations. The PI-directed positive MS/MS spectrum of this component showed an [M + H]+ ion at m/z 572, suggesting a GSH adduct with attachment of a glutathionyl group to nevirapine. The MS/MS spectrum of [M + H]+ ion at m/z 572 provided a characteristic product ion at m/z 443, resulting from neutral loss of pyroglutamate (129 Da) (Fig. 2B). Double neutral losses of glycine and pyroglutamate formed the product ion at m/z 368. The ion at m/z 299 was formed via cleavage of sulfur-carbon bond of the glutathionyl moiety. Further fragmentation of the ion at m/z 299 afforded several fragment ions including ions at m/z 161 and 259 (Fig. 2C). The fragment ion at m/z 259 was formed from the ion at m/z 299 after loss of elements of the cyclopropyl moiety. Formation of the nevirapine GSH adduct constituted approximately 2% of nevirapine and its related metabolites based on the UV chromatogram (Supplemental Fig. S1).
To confirm the structural identity of the nevirapine GSH adduct, 12-trideutero-nevirapine was incubated with HLMs under the same conditions, and the samples were analyzed by LC-MS/MS. As shown in Fig. 3A, only one component was detected by the negative precursor ion scanning of m/z 272. This peak was eluted at approximately 19.7 min and was not detected when either 12-trideutero-nevirapine or NADPH was absent from the incubations. The PI-directed positive MS/MS spectrum of this component showed an [M + H]+ ion at m/z 574. The MS/MS spectrum of [M + H]+ ion at m/z 574 afforded a characteristic product ion at m/z 445, resulting from neutral loss of pyroglutamate (129 Da) (Fig. 3B). The ion at m/z 301 was formed via cleavage of sulfur-carbon bond of the glutathionyl moiety. Further fragmentation of the ion at m/z 301 afforded several fragment ions including ions at m/z 161 and 261 (Fig. 3C). These data suggested that the component is a GSH adduct with attachment of a glutathionyl group to the 4-deuterated methyl group of nevirapine. Glutathione adduction at aryl positions of nevirapine would provide an [M + H]+ ion at m/z 575.
GSH Adduct Formation with Recombinant P450s. To investigate the roles of individual human P450 isozymes in the bioactivation of nevirapine, formation of the nevirapine GSH adduct was examined in incubations of nevirapine with insect cell-expressed recombinant P450s. As shown in Fig. 4, at the same enzyme concentration (100 pmol/ml), CYP3A4 was the predominant enzyme for the formation of the nevirapine GSH adduct. CYP2D6 also catalyzed the formation, but the level was approximately 20% of that formed by CYP3A4. Only minimal amounts or no adduct were detected in incubations with other P450 enzymes including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2E1.

Formation of the NVP GSH adduct in incubations with cDNA-expressed recombinant P450 isozymes. The enzyme activities were expressed as the percentage of CYP3A4 activity and shown as an average of three measurements.
CYP3A4 Inactivation Kinetics of Nevirapine. Preincubation of nevirapine with HLM showed that CYP3A4 was inactivated in a time- and concentration-dependent manner (Fig. 5A). The observed first-order rate constants (kobs) of the inactivation reaction, at a specific nevirapine concentration, were calculated from the slopes of these lines. The hyperbolic plot of kobs versus nevirapine concentrations was shown in Fig. 5B, from which rate constants were obtained. The kinact was determined to be 0.029 min–1 and KI was 31 μM, respectively.
Discussion
The results from the current investigation constitute the first report on the reactive intermediate(s) of nevirapine trapped by the tripeptide nucleophile GSH. Very recently, 12-hydroxynevirapine has been suggested as a mediator of nevirapine-induced hepatotoxicity and skin rash (Antunes et al., 2008; Chen et al., 2008). 12-Hydroxynevirapine has the potential to be sulfated followed by loss of sulfate to form a reactive quinone methide (Fig. 1). The reactivity of 12-sulfoxynevirapine was demonstrated by formation of several DNA adducts in the reactions with 12-mesyloxynevirapine (Antunes et al., 2008). On the other hand, a recent clinical study showed that there was no association between nevirapine-induced toxicities and higher blood levels of 12-hydroxynevirapine in patients (Hall and Macgregor, 2007). To date, 12-sulfoxynevirapine has not been detected in humans and disposition of such a phase II metabolite, if formed in humans, remains unknown. (Riska et al., 1999). In the current study, we demonstrated that formation of the nevirapine GSH adduct occurred in the absence of phase II metabolism like sulfation in human liver microsomal incubations. As suggested by previous studies (Antunes et al., 2008; Chen et al., 2008), the adduct was formed possibly via a dehydrogenation mechanism to generate a quinone methide species. Although the structural identity of the formed nevirapine GSH adduct was supported by LC-MS/MS analysis of the 12-trideutero-nevirapine GSH adduct, in which one methyl deuterium was replaced by attachment of a glutathionyl group (Fig. 3), the absolute structure of the nevirapine GSH adduct was not unequivocally elucidated in this study because the conjugation at an aryl position via a quinone methide species can also result in loss of one deuterium atom. Meanwhile, it is noteworthy to point out that the 12-sulfoxynevirapine pathway can also lead to formation of the quinone methide species (Fig. 1), suggesting a potential bioactivation pathway in vivo. Both pathways share the same initial step of hydrogen atom abstraction of the 4-methyl group of nevirapine. In addition, no other GSH adducts were trapped, suggesting that other metabolic pathways such as 2-, 3-, and 8-hydroxylation are not involved in nevirapine bioactivation.
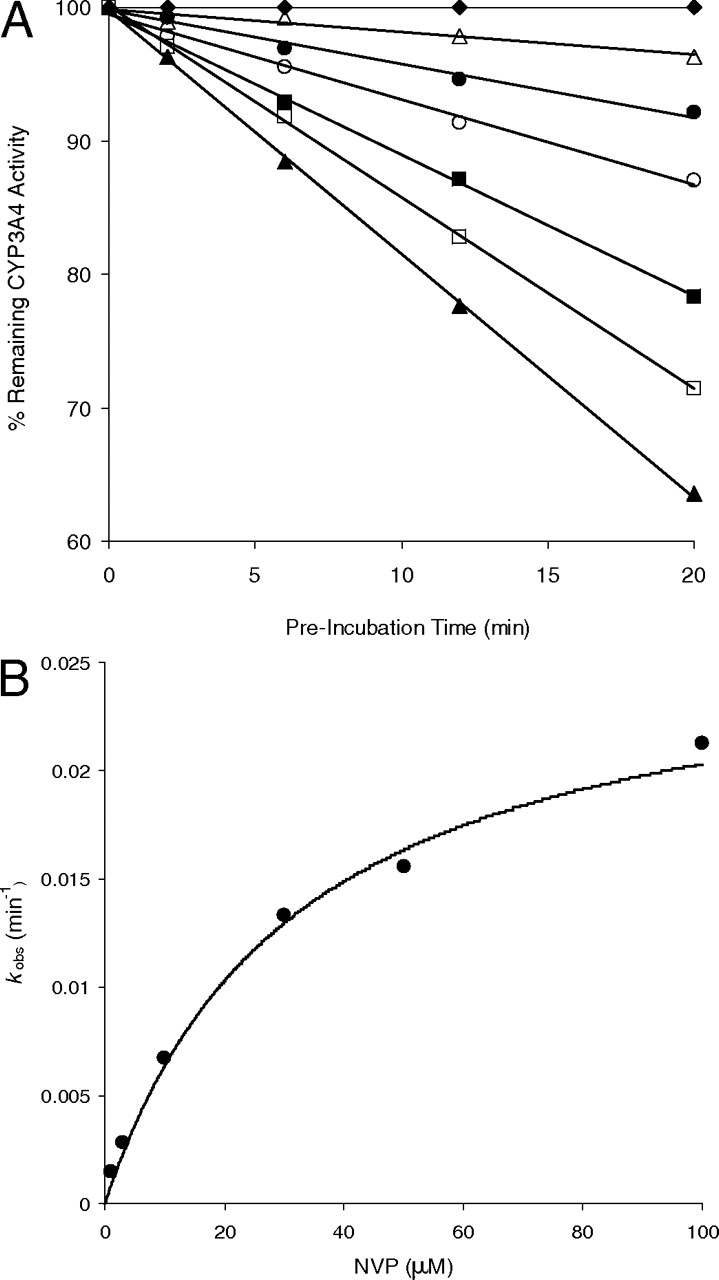
A, time- and concentration-dependent inactivation of CYP3A4 by NVP. (♦), 0 μM; (▵), 1 μM; (•), 3 μM; (○), 10 μM; (▪), 30 μM; (□), 50 μM; and (▴), 100 μM. B, the hyperbolic plot of kobs versus NVP concentrations in HLM.
Apart from the literature report on the involvement of CYP3A4 and CYP2D6 in the 12-hydroxylation of nevirapine (Erickson et al., 1999), our study demonstrated major roles for these enzymes in the metabolic activation of nevirapine. This correlation between 12-hydroxylation and GSH adduct formation can be, at least partially, explained by the fact that the bioactivation pathways via dehydrogenation and formation of 12-hydroxynevirapine share the same initial step of hydrogen atom abstraction of the 4-methyl group (Fig. 1). Inactivation of CYP3A4 by nevirapine was found to be both time- and concentration-dependent with KI = 31 μM and kinact = 0.029 min–1. Such mechanism-based enzyme inactivation was also observed in several HIV protease inhibitors (Ernest et al., 2005).
By trapping the putative reactive intermediate with glutathione, the current study provides direct evidence for the bioactivation of nevirapine. Our studies support the hypothesis that nevirapine undergoes dehydrogenation to form a quinone methide intermediate, which in turn inactivates enzymes conducting hydrogen atom abstraction of the 4-methyl group. These findings are significant for understanding the biochemical mechanisms of idiosyncratic toxicity of the reverse transcriptase inhibitor nevirapine.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.024851.
-
ABBREVIATIONS: NVP, nevirapine; GSH, glutathione; DNVP, 12-trideutero-nevirapine; DMSO-d6, (dimethyl sulfoxide)-D6; HLM, human liver microsome; P450, cytochrome P450; LC-MS/MS, liquid chromatography-tandem mass spectrometry; PI, precursor ion; EPI, enhanced production; HPLC, high-performance liquid chromatography.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Accepted April 10, 2009.
- Received September 25, 2008.
- The American Society for Pharmacology and Experimental Therapeutics






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}