


Abstract
Laromustine (VNP40101M, also known as Cloretazine) is a novel sulfonylhydrazine alkylating (anticancer) agent. Laromustine generates two types of reactive intermediates: 90CE and methylisocyanate. When incubated with rat, dog, monkey, and human liver microsomes, [14C]laromustine was converted to 90CE (C-8) and seven other radioactive components (C-1–C-7). There was little difference in the metabolite profile among the species examined, in part because the formation of most components (C-1–C-6 and 90CE) did not require NADPH but involved decomposition and/or hydrolysis. The exception was C-7, a hydroxylated metabolite, largely formed by CYP2B6 and CYP3A4/5. Laromustine caused direct inhibition of CYP2B6 and CYP3A4/5 (the two enzymes involved in C-7 formation) as well as of CYP2C19. Ki values were 125 μM for CYP2B6, 297 μM for CYP3A4/5, and 349 μM for CYP2C19 and were greater than the average clinical plasma Cmax of laromustine (25 μM). There was evidence of time-dependent inhibition of CYP1A2, CYP2B6, and CYP3A4/5. Treatment of primary cultures of human hepatocytes with up to 100 μM laromustine did not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4/5, but the highest concentration of laromustine decreased the activity and levels of immunoreactive CYP3A4. The results of this study suggest the laromustine has 1) negligible victim potential with respect to metabolism by cytochrome P450 enzymes, 2) negligible enzyme-inducing potential, and 3) the potential in some cases to cause inhibition of CYP2B6, CYP3A4, and possibly CYP2C19 during and shortly after the duration of intravenous administration of this anticancer drug, but the clinical effects of such interactions are likely to be insignificant.
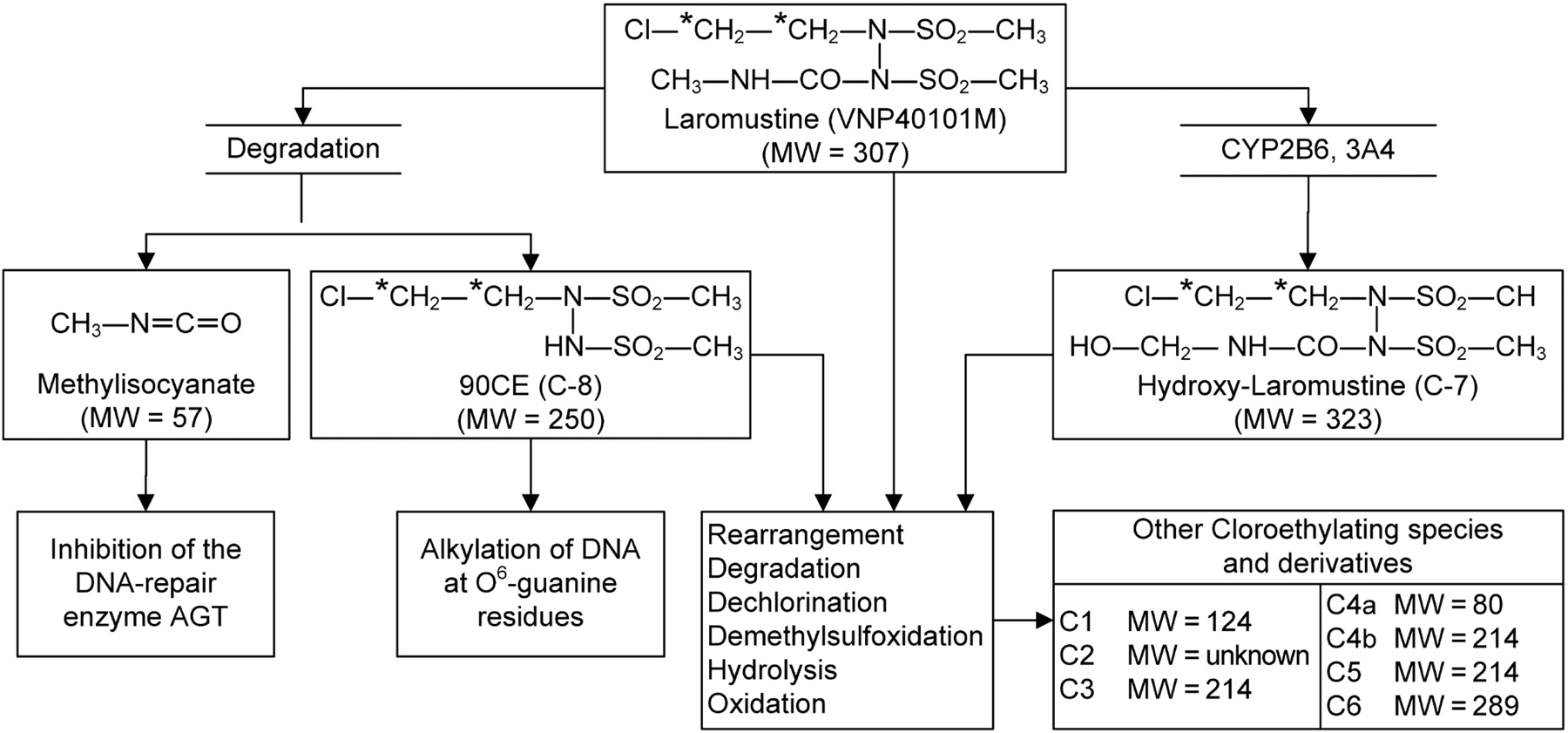
Laromustine [VNP40101M, 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-(methylamino)carbonylhydrazine, also known as Cloretazine] is an active member of a relatively new class of sulfonylhydrazine prodrugs under development as antineoplastic alkylating agents (Penketh et al., 2004; Baumann et al., 2005). As shown in Fig. 1, laromustine generates two distinct types of reactive intermediates: 90CE and methylisocyanate. 90CE and other hard chloroethylating (DNA-reactive) species alkylate DNA at the O6-position of guanine residues that progress to G-C interstrand cross-links (Penketh et al., 2000, 2004). Methylisocyanate, a soft electrophilic carbamoylating agent, binds preferentially and stoichiometrically to sulfhydryl groups and inhibits a number of enzymes, including O6-alkylguanine-DNA-alkyltransferase (AGT), a DNA repair enzyme. The release of methylisocyanate and its inhibition of DNA repair by AGT is thought to augment the antineoplastic effect of 90CE and related chloroethylating species. This process probably accounts for the considerably greater antineoplastic activity of laromustine relative to that of other sulfonylhydrazine alkylating agents, many of which generate 90CE like laromustine, but in contrast to laromustine do not generate methylisocyanate (Baumann et al., 2005).
Laromustine displays anticancer activity against a broad spectrum of transplanted tumors (Finch et al., 2001) and exhibited antileukemic activity in initial clinical trials (Giles et al., 2004). Laromustine is being evaluated in clinical trials for the treatment of acute myelogenous leukemia and bone marrow transplant in patients with cancer.
Drug interactions in patients receiving multiple drug treatments are an important cause of adverse events, as evidenced by the recent withdrawal of newly marketed drugs with unacceptable drug-interaction profiles. Drugs can be withdrawn from the market, denied regulatory approval, or discontinued from clinical development because of their victim potential, which means they are the object of a drug-drug interaction, or their perpetrator potential, which means they are the precipitating cause of the drug interaction (Nassar et al., 2007; Ogilvie et al., 2008).

Chemical structure of laromustine and its metabolites/degradation products.
Victim drugs include terfenadine (Seldane), astemizole (Hismanal), and cisapride (Propulsid), all of which are extensively metabolized by CYP3A4 and can cause torsade de pointes (ventricular tachycardia) when coadministered with drugs that inhibit CYP3A4, such as ketoconazole and erythromycin. Drugs that are extensively metabolized by a polymorphically expressed enzyme, such as CYP2D6, also can be considered victim drugs. Perhexiline and debrisoquine are two drugs that did not receive regulatory approval by the FDA largely because of adverse effects observed in individuals who were genetically deficient in CYP2D6 and, hence, poor metabolizers of these drugs.
Perpetrators are drugs that inhibit or induce drug-metabolizing enzymes and/or drug-transporting proteins. Mibefradil (Posicor) was withdrawn from the U.S. market because of its ability to cause metabolism-dependent (irreversible) inhibition of CYP3A4, resulting in a marked and prolonged inhibition of this important drug-metabolizing enzyme. In contrast to inhibition, induction of drug-metabolizing enzymes may cause a decrease in the plasma concentration of a coadministered drug that may compromise drug efficacy. For example, rifampin (Rifadin), an inducer of CYP3A4 and other drug-metabolizing enzymes, can increase the clearance of several human immunodeficiency viral drugs, cyclosporin A, oral contraceptive steroids, and warfarin, thereby decreasing their therapeutic effectiveness (Parkinson and Ogilvie, 2008).
In this study, we report the results of several in vitro studies that were designed to evaluate the potential for laromustine to be the victim or perpetrator of clinically significant drug interactions. Victim potential was evaluated by comparing the metabolite profile of [14C]laromustine in rat, dog, monkey, and human liver microsomes and by identifying the human cytochrome P450 (P450) enzyme or enzymes that play a role in the metabolism of laromustine. Perpetrator potential was evaluated by assessing the ability of laromustine to inhibit the major P450 enzymes in human liver microsomes and to induce the expression of P450 enzymes in primary cultures of human hepatocytes. These studies were conducted in accordance with the FDA's draft guidance document on the conduct of in vitro metabolism studies (Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis and Implications for Dosing and Labeling, 2006; http://www.fda.gov/cder/guidance/6695dft.pdf) and the principles promulgated by Tucker et al. (2001), Bjornsson et al. (2003), and Huang et al. (2008).
Materials and Methods
Chemicals. Laromustine and [14C]laromustine (labeled in the chloroethylene group) were synthesized at Moravek Biochemicals (Brea, CA). Individual and pooled human liver microsomes (n = 16 or n = 50, mixed gender) were prepared and characterized at XenoTech, LLC (Lenexa, KS), as were Sprague-Dawley rat liver microsomes from a pool of 400 males, beagle dog liver microsomes from a pool of 14 males, and cynomolgus monkey liver microsomes from a pool of 15 males. Recombinant human P450 enzymes expressed in Escherichia coli (Bactosomes) and membranes from E. coli transfected with a plasmid expressing no human enzymes or only human NADPH-cytochrome P450 reductase (without any human P450 enzyme) were obtained from Cypex, Ltd. (Dundee, Scotland). Recombinant human P450 enzymes coexpressed with both NADPH-cytochrome P450 reductase and cytochrome b5 (Supersomes), recombinant human FMO3, and the corresponding control microsomes (from insect cells expressing the ancillary enzymes, but no human P450 enzyme) were purchased from BD Biosciences (San Jose, CA). Rabbit antiserum against human CYP3A4 suitable for Western immunoblotting was obtained from Nosan Corporation (Yokohama, Japan). The secondary antibody (alkaline phosphatase-conjugated goat anti-rabbit IgG) and alkaline phosphatase substrate were purchased from Kirkegaard and Perry Laboratories (Gaithersburg, MD). All other reagents were obtained from commercial sources, most of which have been described elsewhere (Robertson et al., 2000; Madan et al., 2003; Ogilvie et al., 2006).
In Vitro Metabolism of [14C]Laromustine. [14C]Laromustine (up to 100 μM) was incubated in duplicate at 37 ± 1°C with pooled human liver microsomes (1 mg of protein/ml) in 0.25-ml incubation mixtures containing potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM, pH 7.4), and a NADPH-generating system consisting of NADP (1 mM, pH 7.4), glucose 6-phosphate (5 mM, pH 7.4), and glucose-6-phosphate dehydrogenase (1 unit/ml), at the final concentrations indicated. Reactions were initiated by the addition of [14C]laromustine (dissolved in DMSO; final solvent concentration 0.1%, v/v) and were terminated after 0 to 60 min by the addition of an equal volume of 20% (v/v) perchloric acid. Precipitated protein was removed by centrifugation (920g for 10 min at 10°C), and the supernatant fractions were analyzed by HPLC with radiometric detection. Calibration standards of [14C]laromustine were used to quantify metabolite formation and loss of parent compound. Zero time, zero substrate, zero protein, and zero cofactor incubations served as controls.
To assess the involvement of individual recombinant human P450 enzymes and FMO3 in metabolite formation, [14C]laromustine (25 and 100 μM) was incubated in duplicate for 10 min at 37 ± 1°C with a panel of recombinant human P450 enzymes (rCYP1A2, rCYP2A6, rCYP2B6, rCYP2C8, rCYP2C9, rCYP2C19, rCYP2D6, and rCYP3A4) at 25 pmol of P450/incubation and with recombinant FMO3 at 1 mg of protein/ml. The incubation conditions and sample workup were similar to those described above.
[14C]Laromustine at a final concentration of 25 μM (the average plasma Cmax value) was incubated with a bank of human liver microsomes (n = 16) to determine the interindividual differences in metabolite (C-7) formation. [14C]Laromustine was incubated in duplicate for 0 and 10 min at 37 ± 1°C with a single concentration of human liver microsomes (1 mg of protein/ml). The incubation conditions and sample workup were as described above. The 16 samples of human liver microsomes used were characterized at XenoTech, LLC to determine the sample-to-sample variation in the activity of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, CYP4A11, and FMO.
In Vitro P450 Inhibition. The ability of laromustine to inhibit the major drug-metabolizing P450 enzymes in a direct and time-dependent manner was investigated with a pool of human liver microsomes (n = 16). The marker substrates, the concentration of microsomal protein and each substrate, incubation times, and incubation conditions were all described by Ogilvie et al. (2006, 2008). Because laromustine is unstable in the presence of human liver microsomes, reactions were initiated by the addition of laromustine to minimize degradation. To evaluate laromustine as a direct-acting inhibitor, pooled human liver microsomes (0.1 mg/ml or less) were incubated with P450 marker substrates at multiple concentrations that bracketed Km (as described in Ogilvie et al., 2008) in the presence and absence of laromustine (at concentrations ranging from 25 to 750 μM) to determine the inhibition constant (Ki) and the mechanism of inhibition.
To assess time-dependent inhibition, laromustine (at a target concentration of 100 μM) was preincubated for 0, 15, or 30 min in the presence and absence of NADPH with human liver microsomes at 0.05 mg/ml for midazolam samples and at 0.1 mg/ml for all other samples or at 25 times these protein concentrations for samples that were subsequently diluted 25-fold before P450 activity was measured. To minimize the direct-acting inhibitory effects of laromustine and its metabolites/degradation products, P450 activity was measured with marker substrates at target concentrations ranging from 2 to 10 times Km as follows: 3 times Km for phenacetin, 10 times Km for bupropion, 2 times Km for testosterone, and 10 times Km for midazolam (Ogilvie et al., 2008).
In Vitro P450 Induction. The ability of laromustine to induce or suppress the expression of P450 enzymes was investigated in primary cultures of freshly isolated human hepatocytes with a Matrigel overlay. After a 2-day adaptation period, three preparations of cultured human hepatocytes from three separate human livers were treated once daily for 3 consecutive days with laromustine (1, 10, and 100 μM) or one of three prototypical P450 inducers, omeprazole (100 μM), phenobarbital (750 μM), and rifampin (10 μM), at the final concentrations indicated. Laromustine and the positive controls were dissolved in DMSO, and hepatocytes treated with DMSO (final concentration 0.1%, v/v) served as negative controls. The isolation, culturing, and treatment procedures were performed essentially as described by Madan et al. (2003). Human hepatocytes were harvested 24 h after the third and final treatment to prepare microsomes, which were analyzed for selected P450 activities with enzyme-selective substrates.
In addition to measuring CYP3A4/5 activity in the microsomal samples, the levels of CYP3A4 were also determined by Western immunoblotting. Microsomes (10 μg/lane) were separated by SDS-polyacrylamide gel electrophoresis based on the method of Laemmli (1970), after which proteins were transferred to a polyvinylidene difluoride membrane based on the method of Towbin et al. (1979) as modified by Arlotto et al. (1989). The polyvinylidene difluoride membranes were incubated overnight at room temperature with rabbit polyclonal anti-CYP3A4 antiserum (1:5000 dilution), followed by incubation for 90 to 120 min with the secondary antibody alkaline phosphatase-conjugated goat anti-rabbit IgG (1:5000). Membranes were developed with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolylphosphate (0.21 mg/ml) and nitroblue tetrazolium (0.41252 mg/ml) to visualize immunoreactive CYP3A4. Blots were air-dried and scanned with an Astra 1200S scanner (UMAX Data Systems, Fremont, CA), and band intensities were quantified with Intelligent Quantifier gel densitometry software (version 3.0; Genomic Solutions, Ann Arbor, MI).
Analytical Methods. [14C]Laromustine and its metabolites/degradation products were analyzed by reverse-phase HPLC with a radioactivity detector. The HPLC system consisted of two Shimadzu LC-10ADvp pumps, a Shimadzu autosampler, and a Shimadzu DGU-14A degasser (Shimadzu, Columbia, MD) with a β-Ram radioactivity detector equipped with a 500-μl UHP LiGl flow cell (IN/US Systems, Tampa, FL). For all experiments, the HPLC column was a Phenomenex Prodigy ODS (5-μm particle size, 250 mm × 4.6 mm) column. Mobile phase A was 5 mM ammonium acetate in water, whereas mobile phase B was 0.1% (v/v) formic acid in methanol. The column was maintained at 40 ± 5°C with a column heater, and the flow rate was 0.15 ml/min from 0 to 30 min and 0.25 ml/min from 30 to 60 min. Both [14C]laromustine and metabolites of interest were quantified based on the radioactivity response of standard solutions of [14C]laromustine.
All analyses of P450 enzyme activities were performed with validated HPLC-tandem mass spectrometry methods as described by Ogilvie et al. (2008). The mass spectrometry equipment was either an ABI Sciex (Applied Biosystems/MDS Sciex, Foster City, CA), API 2000, API 3000, or an API 4000 mass spectrometer with Shimadzu HPLC pumps and autosampler systems. Metabolites were quantified by back calculation of a weighted (1/x), linear, least-squares regression. The regression fit was based on analyte/internal standard peak area ratios calculated from calibration standard samples, which were prepared from authentic metabolite standards. Peak areas were integrated with an Applied Biosystems/MDS Sciex Analyst data system (version 1.3.1 or later).
Statistical Tests and Data Processing. In vitro metabolism data were processed with the spreadsheet computer program Microsoft Excel (Microsoft Excel 2003; Microsoft, Redmond, WA). The parent compound and metabolites were both quantified from calibration curves based on replicates of six concentrations of the parent compound, which assumes that metabolites and parent compound have the same radiometric response. Data were processed with Analyst 1.4 system software. Correlation analysis was performed with the computer software SigmaStat (version 2.03; Systat Software, Inc., San Jose, CA).
P450 inhibition data were processed with Microsoft Excel (Office 2000 version 9.0; Microsoft). For all Ki determinations, the entire data set (i.e., reaction rates at all concentrations of laromustine and marker substrate) were fitted to Michaelis-Menten equations for competitive, noncompetitive, uncompetitive, and mixed (competitive-noncompetitive) inhibition by nonlinear regression analysis with GraFit (version 4.0.21; Erithacus Software, Horley, Surrey, UK). Goodness of fit (χ2 analysis) applied to the equations for competitive, noncompetitive, uncompetitive, and mixed inhibition was used to select the type of inhibition observed. Data on the effects of NADPH and protein dilution were processed with a customized add-in for the computer program Microsoft Excel (Office 2000 version 9.0). Calculations to estimate the concentration of laromustine supporting half the maximum rate of inactivation (KI) and kinact values were based on methods described by Kitz and Wilson (1962). These estimates assume that the rate of inactivation is linear between 0 and 30 min and provide a lower limit for the kinact value.
P450 induction data were processed with Microsoft Excel (Office 2000 version 9.0). To assess the statistical significance of differences between group means, equal variance and normality tests were conducted first to determine whether the data were parametrically distributed. Subsequently, for parametrically distributed data, a one-way repeated-measures analysis of variance (ANOVA) was performed to identify significant differences between group means. For nonparametrically distributed data, a Kruskal-Wallis ANOVA was performed. The ANOVA was followed by a Dunnett's post hoc test to identify group means that were significantly different from control (p < 0.05 or 5% level of significance). This statistical test is designed to make multiple comparisons with a single mean, such as comparing multiple treatment groups with a single control group. Statistical analyses were performed with a SigmaStat statistical analysis system (version 2.03; SPSS Inc., Chicago IL).
Results
In Vitro Metabolism of [14C]Laromustine. Laromustine readily undergoes base-catalyzed (nonenzymatic) conversion to methylisocyanate and 90CE, which, in turn, degrades to other chloroethylating derivatives, as shown in Fig. 1. The rate of disappearance of [14C]laromustine (100 μM) in the presence of NADPH-fortified human liver microsomes was essentially the same as the rate of laromustine loss in the absence of NADPH or the presence of boiled (heat-denatured) microsomes and was the same as the nonenzymatic rate of laromustine degradation in potassium phosphate buffer. These results suggest that human liver microsomal P450 enzymes and carboxylesterases do not contribute substantially to the overall rate of laromustine disappearance.
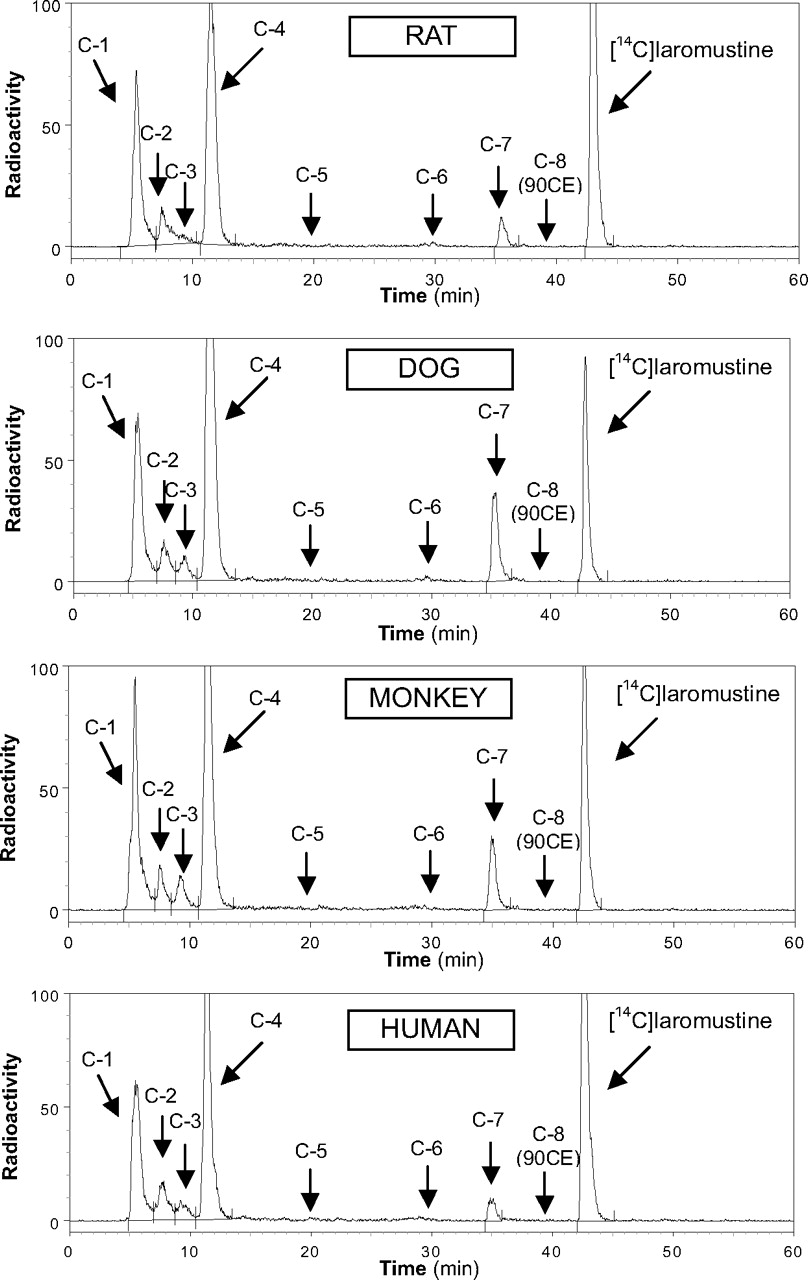
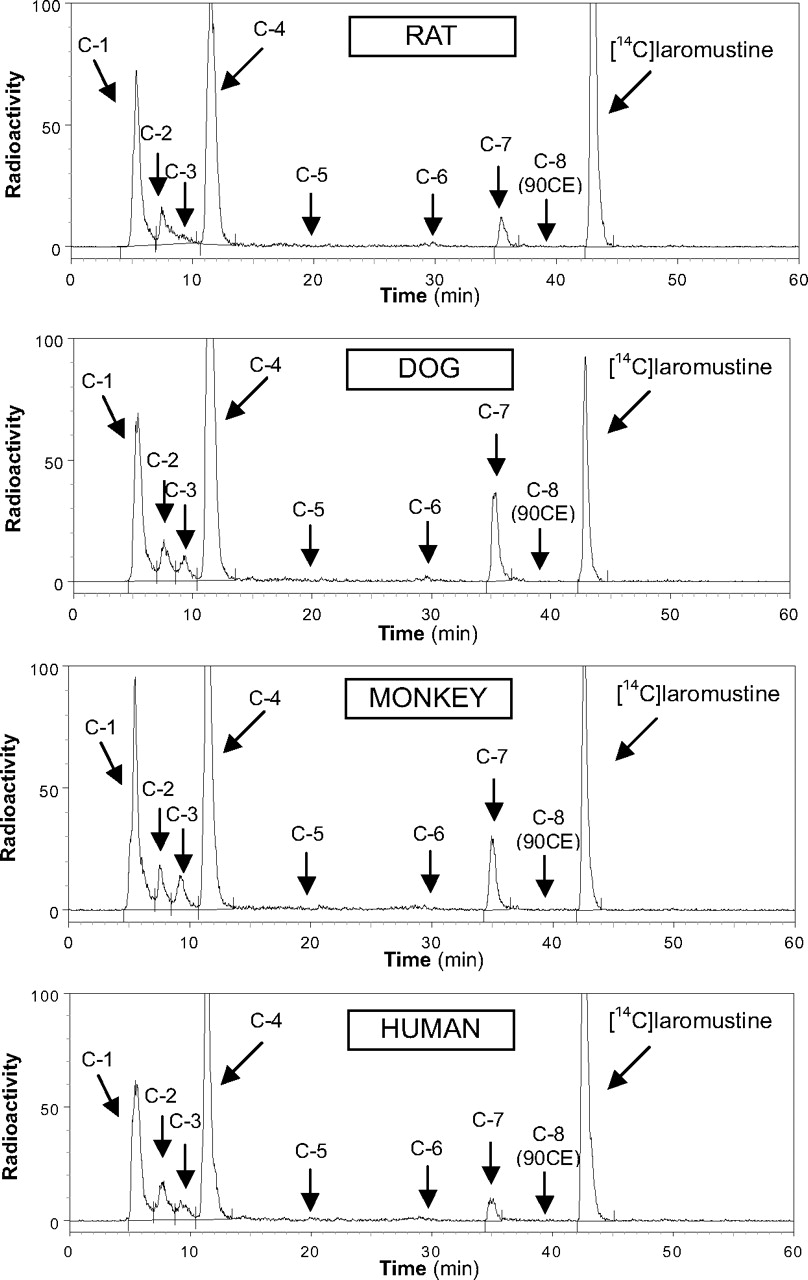
Chromatograms from incubations of [14C]laromustine (100 μM) with rat, dog, monkey, and human liver microsomes in the presence of NADPH.
Similar results were obtained with NADPH-fortified rat liver microsomes. However, the rate of laromustine disappearance was enhanced over the rate of nonenzymatic degradation when [14C]laromustine (100 μM) was incubated with NADPH-fortified liver microsomes from cynomolgus monkeys or beagle dogs. These results suggest that dog and monkey liver microsomes contain P450 enzymes (or other NADPH-dependent enzymes) that metabolize laromustine at a substantial rate, compared with the nonenzymatic rate of laromustine degradation, whereas rat and human P450 enzymes do not. This interpretation was supported by the results of metabolite profiling.
The in vitro profile of [14C]laromustine metabolites/degradation products formed by liver microsomes from rats, dogs, monkeys, and humans is shown in Fig. 2. When [14C]laromustine (100 μM) was incubated with rat, dog, monkey, and human liver microsomes in the presence of NADPH, eight radioactive components (C-1 through C-8) were detected, and five of them (C-1, C-2, C-3, C-4, and C-7) were detected after 60 min of incubation (Fig. 2). The metabolite profile was similar for all species with two notable exceptions: first, C-3 was either not formed or formed at low levels with rat liver microsomes, and second, C-7 was a considerably more prominent component in dog and monkey liver microsomes than in rat and human liver microsomes. Component C-8 cochromatographed (retention time ∼39 min) with an authentic standard of 90CE; it was a major component at early time points and at low substrate concentration regardless of the source of liver microsomes (results not shown). Formation of 90CE (C-8) is associated with formation of methylisocyanate, but formation of methylisocyanate from [14C]laromustine was not detected by radiometric HPLC because it lacks the 14C label, which is part of the chloroethyl moiety, as shown in Fig. 1.
Of the radioactive components detected, only the formation of C-7 was dependent on NADPH. Formation of C-7 involved methyl hydroxylation of laromustine (+16 atomic mass unit) on the methylisocyanate moiety (A. Nassar, I. King, B. Paris, L. Haupt, F. Ndikum-Moffor, R. Campbell, E. Usuki, J. Skibbe, D. Brobst, B. Ogilvie, et al., manuscript in preparation). The time course of C-7 formation by liver microsomes from each species indicates that once formed, C-7 is further metabolized or degraded. The rate of formation of C-7 followed the rank order (from fastest to slowest): dog ≈ monkey > rat ≈ human, which corresponds to the same rank order for the overall rate of laromustine disappearance.
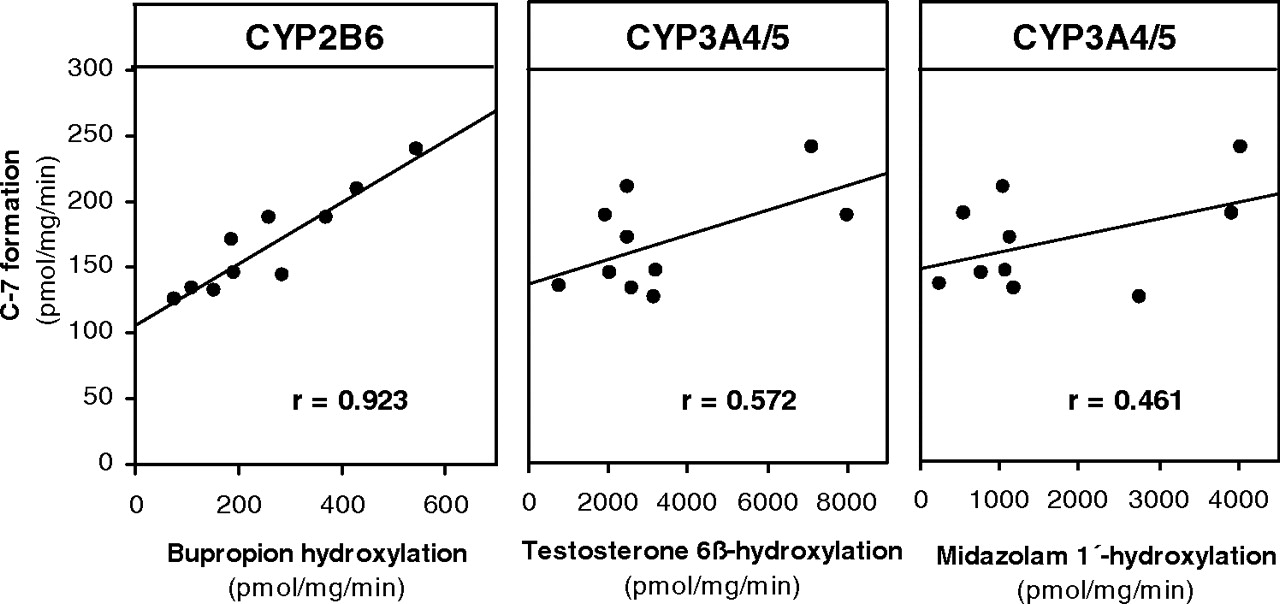
Correlation between the rate of formation of C-7 from [14C]laromustine (25 μM) and CYP2B6 and CYP3A4/5 activity in a bank of human liver microsomes (n = 10).
To identify the human P450 enzyme or enzymes responsible for C-7 formation, laromustine (25–100 μM) was incubated with a bank (n = 10) of individual samples of human liver microsomes (correlation analysis) and a panel of recombinant human enzymes. Of the recombinant human enzymes evaluated, only CYP2B6 and CYP3A4/5 converted laromustine to C-7 and did so at comparable rates. With the recombinant P450 enzymes, C-7 formation was evaluated at a single time point (10 min) based on time course experiments with human liver microsomes. We cannot exclude the possibility that one or more recombinant P450 enzymes formed C-7 at a later or earlier time point. As shown in Fig. 3, the sample-to-sample variation in the conversion of [14C]laromustine (25 μM) to C-7 correlated strongly (r = 0.923) with CYP2B6 (bupropion hydroxylase) activity. It also correlated weakly with CYP3A4/5 activity based on testosterone 6β-hydroxylation (r = 0.572) and midazolam 1′-hydroxylation (r = 0.461). The linear regression lines for CYP2B6 and CYP3A4/5 did not pass through or near the origin, suggesting that both enzymes are involved in the formation of C-7. The correlation between C-7 formation and CYP2B6 activity (r = 0.923) improved when the variation in CYP3A4/5 activity was also taken into consideration (r = 0.945).
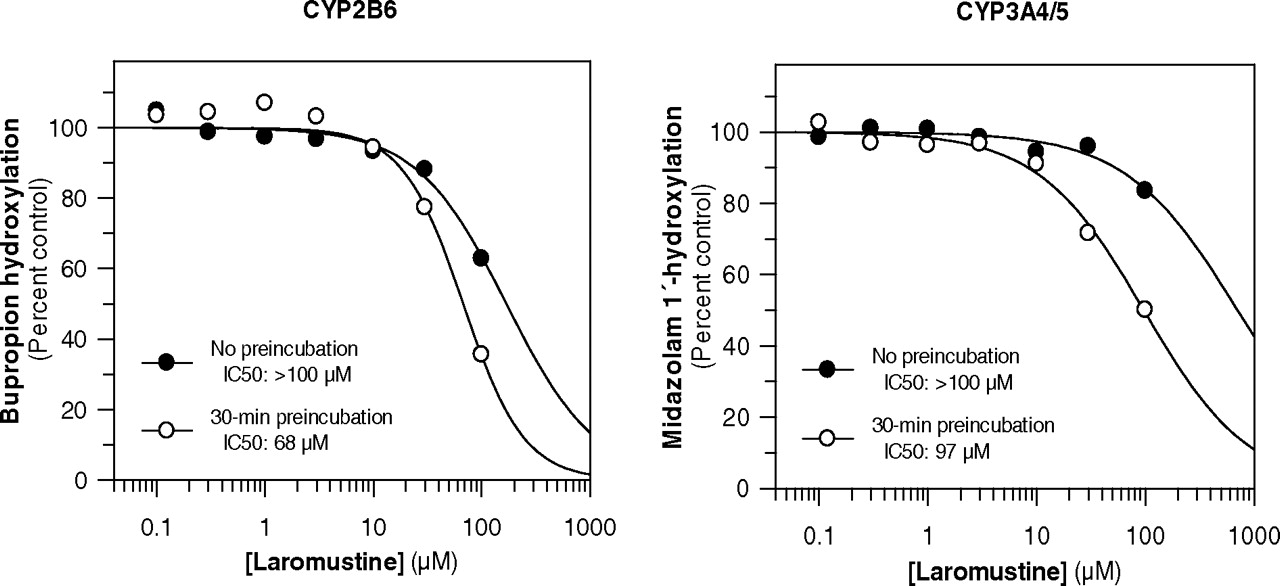
In Vitro P450 Inhibition. Laromustine was evaluated as an inhibitor of several P450 enzymes both before and after a 30-min preincubation with NADPH-fortified human liver microsomes to examine the potential for direct inhibition and time-dependent inhibition. As shown in Fig. 4, laromustine caused time-dependent inhibition of CYP2B6 (bupropion hydroxylation) and CYP3A4/5 (midazolam 1′-hydroxylation), as evidenced by a time-dependent shift in IC50 curves. There was some evidence of time-dependent inhibition of CYP1A2, but the degree of inhibition at all concentrations and time points was less than 50% (results not shown). Time-dependent inhibition of CYP1A2, CYP2B6, and CYP3A4/5 by laromustine was not dependent on the presence of NADPH, although the presence of NADPH slightly enhanced the time-dependent inhibition of CYP2B6 and CYP3A4/5 (results not shown). When these experiments were repeated with a 25-fold higher concentration of human liver microsomes (i.e., 2.5 mg/ml) followed by a 25-fold dilution before P450 activity was measured, the time-dependent inhibition of CYP1A2, CYP2B6, and CYP3A4/5 was partially, but not completely, reversed. This finding suggests that time-dependent inhibition of these enzymes involves a combination of enzyme inactivation and the formation of metabolites/degradation products that are more potent than laromustine as direct-acting (reversible) P450 inhibitors.
For the time-dependent inhibition of CYP3A4/5, KI was estimated to be 83 μM, and kinact was estimated to be 0.032 min–1, indicating that 3.2% of CYP3A4/5 is inactivated per minute in the presence of saturating concentrations of laromustine (results not shown). Based on kinact/KI, the intrinsic efficiency of CYP3A4/5 inactivation by laromustine (0.39 min–1 mM–1) was greater than that for CYP2B6, which is apparent from the relatively small shift in IC50 curves for CYP2B6, compared with those for CYP3A4/5 (Fig. 4).
To determine the inhibition constant Ki and mechanism of inhibition of the major human P450 enzymes involved in drug metabolism, human liver microsomes (pool of 16) were incubated with P450 marker substrates at multiple concentrations of Km/3, Km, 3 times Km, 6 times Km, and 10 times Km in most, but not all, cases and with a wide range of laromustine concentrations (25–750 μM). As shown in Table 1, the highest concentration of laromustine evaluated (750 μM) caused less than 25% inhibition of CYP1A2, CYP2C8, CYP2C9, and CYP2D6 at marker substrate concentrations equal to Km. Even at marker substrate concentrations of 0.25 or 0.3 Km, the highest concentration of laromustine caused less than 50% inhibition of these P450 enzymes, indicating that the Ki values for inhibition of CYP1A2, CYP2C8, CYP2C9, and CYP2D6 are greater than 750 μM. Eadie-Hofstee plots for CYP1A2, CYP2C8, CYP2C9, and CYP2D6 were characterized by a series of parallel lines consistent with noncompetitive inhibition (results not shown). There was no evidence of time-dependent inhibition of these enzymes, with the exception of CYP1A2 (as mentioned above).
Evaluation of laromustine as a direct-acting inhibitor of selected P450 enzymes in human liver microsomes
As shown in Fig. 5 and Table 1, laromustine inhibited CYP2B6 and CYP3A4/5, the two enzymes implicated in the metabolism of laromustine to C-7, as well as CYP2C19. With no preincubation, laromustine appeared to cause competitive inhibition of CYP2B6 (bupropion hydroxylation) with a Ki value of 125 μM and caused mixed (competitive-noncompetitive) inhibition of CYP3A4/5 (midazolam 1′-hydroxylation) and CYP2C19 (S-mephenytoin 4′-hydroxylation) with Ki values of 297 and 349 μM, respectively (Fig. 5). It is interesting to note that the inhibition of CYP3A4/5 was substrate-dependent, inasmuch as laromustine inhibited the 1′-hydroxylation of midazolam to a greater extent than it inhibited the 6β-hydroxylation of testosterone. Laromustine caused less than 50% inhibition of testosterone 6β-hydroxylation even at a marker substrate concentration of 0.3 Km (i.e., 30 μM testosterone).

Effect of preincubation of laromustine with human liver microsomes and NADPH on the inhibition of CYP2B6 (left) and CYP3A4/5 (right).
In Vitro P450 Induction. To evaluate the enzyme-inducing potential of laromustine, three preparations of cultured human hepatocytes were treated once daily for 3 consecutive days with laromustine (1, 10, or 100 μM) or one of three prototypical enzyme inducers, omeprazole (100 μM), phenobarbital (750 μM), and rifampin (10 μM), used as positive controls. Microsomes were prepared 24 h after the final treatment and assayed for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4/5 activity. Under the conditions examined, laromustine caused no cell toxicity based on light microscopic evaluation. Throughout the treatment period, hepatocytes remained cuboidal in shape with intact cell membranes and granular cytoplasm, and they maintained a confluent monolayer with few intercellular spaces.
As shown in Fig. 6, all three preparations of human hepatocytes responded as expected to treatment with the prototypical P450 inducers: Treatment with omeprazole caused a marked increase in CYP1A2 (∼22-fold), whereas treatment with phenobarbital or rifampin caused an increase in CYP2B6 (5–10-fold), CYP2C8 (8–12-fold), CYP2C9 (5–6-fold), CYP2C19 (4–7-fold), and CYP3A4/5 (10–12-fold). Treatment of human hepatocytes for 3 consecutive days with up to 100 μM laromustine had little or no effect on CYP1A2, CYP2B6, CYP2C8, CYP2C9, or CYP2C19 activity. However, at the highest concentration tested (100 μM), laromustine caused a 41% decrease in CYP3A4/5 activity, which was associated with a corresponding decrease (∼70%) in the levels of immunoreactive CYP3A4, as shown in Fig. 7.
Discussion
For a drug whose clearance is dependent on hepatic metabolism, victim potential can be assessed on the basis of the fractional metabolism (fm) of the drug by a given P450 or other drug-metabolizing enzyme (Ogilvie et al., 2008). Fractional metabolism determines the increase in systemic exposure to the drug when a given P450 enzyme is inhibited or genetically deficient, according to the following equation:  where AUC is area under the curve.
where AUC is area under the curve.
When fm = 0.2 (i.e., when a given P450 enzyme accounts for 20% of the clearance of a drug), loss of that P450 enzyme will result in a 1.25-fold increase in AUC. Therefore, the inhibition or genetic loss of a P450 enzyme that contributes 20% or less to the clearance of a drug will not affect systemic exposure to a drug beyond the 80 to 125% window of equivalence. Presumably for this reason, the FDA requires the characterization of all metabolic pathways that contribute 25% or more (fm ≥ 0.25) to a drug's clearance (Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis and Implications for Dosing and Labeling, 2006; http://www.fda.gov/cder/guidance/6695dft.pdf).

Eadie-Hofstee plots (Ki determination) for the inhibition of CYP2B6, CYP2C19, and CYP3A4/5 by laromustine.
The present study was undertaken to determine whether one or more P450 enzymes in human liver microsomes can potentially contribute 25% or more to the clearance of laromustine, an anticancer drug that undergoes base-catalyzed conversion to 90CE and other chloroethylating species that alkylate DNA in the O6-position of guanine residues, and methylisocyanate, which inhibits the repair of O6-alkylated residues by AGT (Penketh et al., 2004; Baumann et al., 2005). Studies with a panel of recombinant human P450 enzymes and correlation analysis with a bank of human liver microsomes implicated CYP2B6 and CYP3A4/5 in the hydroxylation of laromustine to C-7, which is one of eight radioactive components detected when [14C]laromustine is incubated with human liver microsomes, and C-7 is the only component whose formation is dependent on NADPH (Fig. 2). However, the results suggest that inhibition of CYP2B6 and CYP3A4/5 will not affect the pharmacokinetics of laromustine, because the rate of disappearance of [14C]laromustine (100 μM) in the presence of NADPH-fortified human liver microsomes was essentially the same as the rate of laromustine loss in the absence of NADPH or the presence of boiled (heat-denatured) microsomes and was the same as the nonenzymatic rate of laromustine degradation in potassium phosphate buffer. Therefore, although CYP2B6 and CYP3A4/5 convert laromustine to C-7, metabolism by P450 (or any other microsomal enzyme) does not contribute substantially to the overall rate of laromustine disappearance, which is largely determined by nonenzymatic degradation.

Effects of treating cultured human hepatocytes with laromustine or prototypical inducers on microsomal P450 activity. For CYP1A2, CYP2C9, and CYP3A4/5, the results are from three preparations of human hepatocytes. Because of the limited amount of microsomal protein, values for CYP2B6 and CYP2C8 are from two preparations, and for CYP2C19 the results are from a single preparation of human hepatocytes.

Effects of treating cultured human hepatocytes with laromustine on the levels of immunoreactive CYP3A4. Microsomes (10 μg) from three preparations of human hepatocytes treated with laromustine (1, 10, or 100 μM) or prototypical enzyme inducers (100 μM omeprazole, 750 μM phenobarbital, or 10 μM rifampin) were analyzed by Western immunoblotting with antibody against CYP3A4. Additional controls included human liver microsomes (HLM) with low, mid, and high CYP3A4 activity and recombinant CYP3A4 (Bactosomes).
Metabolism by P450 did enhance the rate of laromustine disappearance in NADPH-fortified liver microsomes from cynomolgus monkeys and beagle dogs but not from humans or rats. These results suggest that P450 enzymes (or other NADPH-dependent enzymes) in dog and monkey liver microsomes hydroxylate laromustine to C-7 at an appreciable rate, compared with the nonenzymatic rate of laromustine degradation, whereas rat and human P450 enzymes do not. Nevertheless, the profile of laromustine metabolites/degradation products was remarkably similar across all species examined (Fig. 2). It is significant that no human-specific in vitro metabolites/degradation products were detected.
To evaluate its perpetrator potential, laromustine was evaluated as an inducer/suppressor of P450 enzymes in cultured human hepatocytes and as a direct-acting and time-dependent inhibitor of P450 enzymes in human liver microsomes. Under conditions in which the positive controls (omeprazole, phenobarbital, and rifampin) caused anticipated induction of the appropriate P450 enzymes, laromustine (1–100 μM) did not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4/5 in the three preparations of cultured human hepatocytes, which suggests that laromustine will not cause clinically significant enzyme induction. However, the highest concentration of laromustine tested (100 μM) caused a decrease in CYP3A4/5 activity and CYP3A4 immunoreactive protein levels. Although the decrease in CYP3A4/5 activity can be linked to the decrease in immunoreactive CYP3A4 levels, it is not known whether laromustine suppressed the expression of CYP3A4/5 or whether it inactivated CYP3A4/5, thereby stimulating its proteolytic degradation. The possible clinical implications of this decrease in CYP3A4/5 levels are discussed below in connection with the inhibition of P450 enzymes by laromustine.
Laromustine caused competitive inhibition of CYP2B6 and a mixed (competitive-noncompetitive) inhibition of CYP2C19 and CYP3A4/5 (as measured by midazolam 1′-hydroxylation) with calculated Ki values of 125, 349, and 297 μM, respectively (Fig. 5). For CYP2B6 and CYP3A4/5, the two enzymes implicated in the conversion of laromustine to C-7, there was also evidence of time-dependent inhibition (Fig. 4). Furthermore, laromustine decreased CYP3A4/5 activity and levels in cultured human hepatocytes (Figs. 6 and 7). All of these results raise the possibility that although laromustine may cause drug-drug interactions by impairing the metabolism of drugs by CYP2B6, CYP2C19, and CYP3A4/5, such interactions are likely to be insignificant.
To assess the potential for laromustine to cause clinically significant P450 inhibition, the in vitro data were extrapolated to the in vivo situation based on the relevant FDA Guidance for Industry (http://www.fda.gov/cder/guidance/6695dft.pdf), which recommends that in vitro to in vivo extrapolations be based on the value of [I]/Ki, where [I] is the maximum concentration of total drug (bound and free) in the systemic circulation at steady state (Cmaxss), and Ki is the inhibition constant determined in vitro. Based on this conservative approach, the FDA categorizes [I]/Ki values in terms of the likelihood of clinically significant P450 inhibition as follows: less than 0.1 = unlikely; more than 1.0 = probable; and values between 0.1 and 1.0 = possible.
As is the case with other anticancer alkylating agents, laromustine is not dosed repeatedly to a steady state but is infused once via a freely flowing peripheral or central intravenous line over a period of 30 to 90 min. Most patients receive the dose over 30 to 35 min in a total volume of 500 ml. A second dose, if given, is not administered until 35 days later. When administered under these conditions at a dose of 600 mg/m2, the average plasma Cmax for laromustine was approximately 25 μM (Vion studies CLI-033b and CLI-043b; unpublished data). The estimated Ki values listed in Table 1 were used to calculate [I]/Ki values for the three most potently inhibited P450 enzymes, and the results are summarized in Table 2. For CYP2B6, the most potently inhibited enzyme, the [I]/Ki value exceeds 0.1 based on an average Cmax of 25 μM and exceeds 1.0 based on the highest Cmax of 128 μM as shown in Table 2 (Vion studies CLI-033b and CLI-043b). For CYP2C19 and CYP3A4/5, [I]/Ki values were less than 0.1 based on the average Cmax value but approximately 0.4, based on the highest Cmax value reported in the two 600 mg/m2 clinical studies (Vion CLI-033b and CLI-043b). Based on the FDA's Guidance document, these [I]/Ki values suggest that laromustine will possibly cause inhibition of CYP2B6, CYP2C19, and CYP3A4/5, especially in patients with high plasma levels of this anticancer drug. However, the approach to estimating the perpetrator potential of laromustine may overestimate the ability of laromustine to cause clinically significant P450 inhibition because the values of [I] are not based on steady-state Cmax values but on the highest concentration of laromustine measured after intravenous administration. Laromustine is infused over a 30- to 90-min period, and blood levels of laromustine are negligible after 4 h (Vion studies CLI-033b and CLI-043b). Therefore, if laromustine does inhibit CYP2B6, CYP2C19, or CYP3A4/5, it would be expected to do so only for a short period immediately after its administration. In addition, laromustine is administered only once, and on those occasions when a second treatment is required, the two drug treatments are separated by at least 35 days. In both protocols CLI-033 CLI-043, the second dose can be given no earlier than 35 days and no later than 60 days.
P450 inhibition by laromustine: estimates of [I]/Ki based on various clinical estimates of plasma Cmax
In addition to causing direct inhibition, laromustine caused time-dependent inhibition of CYP3A4/5 and, to a lesser extent, CYP2B6, and at the highest concentration tested (100 μM) laromustine decreased the levels of CYP3A4/5 in human hepatocytes. Based on kinact/KI, the intrinsic efficiency of CYP3A4/5 inactivation by laromustine was estimated to be 0.39 min–1 mM–1. In a recent review article, Jones et al. (2007) reported that kinact/KI values for metabolism-dependent inhibitors of CYP3A4 ranged from values in the thousands for drugs such as troleandomycin (1222), mifepristone (1634), saquinavir (1824), amprenavir (2808), and ritonavir (3200) to values in the teens for drugs such as N-desmethyltamoxifen (11), fluvoxamine (13), fluoxetine (17), and nortriptyline (19). Therefore, relative to other metabolism-dependent inhibitors, laromustine has very low intrinsic efficiency for inactivating CYP3A4. Based on its low intrinsic efficiency and the fact that laromustine is administered once, it seems reasonable to conclude that laromustine will not inactivate CYP3A4/5 to a clinically significant extent.
Patients with cancer always receive multiple drugs, including chemotherapeutic agents and antibiotics for the management of diseases. It is therefore important to investigate the potential pharmacokinetic or pharmacodynamic interactions between drugs. The results of this study suggest laromustine has 1) negligible victim potential with respect to metabolism by P450 enzymes, 2) negligible enzyme-inducing potential, and 3) the potential in some cases to cause inhibition of CYP2B6, CYP3A4, and possibly CYP2C19 during and shortly after the duration of intravenous administration of this anticancer drug, but the clinical effects of such interactions are likely to be insignificant.
Footnotes
-
This work was supported by Vion Pharmaceuticals, Inc., New Haven, CT.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.027516.
-
ABBREVIATIONS: VNP40101M, 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-(methylamino)carbonylhydrazine, laromustine, also known as Cloretazine; 90CE, a pharmacologically active degradation product of laromustine (as VNP4090CE and, in this study, C-8); AGT, O6-alkylguanine-DNA-alkyltransferase; FDA, Food and Drug Administration; P450, cytochrome P450; FMO, flavin-containing monooxygenase; DMSO, dimethyl sulfoxide; HPLC, high-performance liquid chromatography; r, recombinant; C-7, VNP40107 (a metabolite of laromustine); ANOVA, analysis of variance.
- Accepted April 8, 2009.
- Received March 10, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}