


Abstract
Volume of distribution (VD) is a key pharmacokinetic property that together with clearance determines the half-life or residence time of drug in the body. It is commonly expressed as steady-state volume of distribution VDss with a physiological basis for its understanding developed by Øie and Tozer in 1979. The Øie-Tozer equation uses terms for plasma protein binding (fup), tissue binding (fut), and the extravascular/intravascular ratio of albumin as well as constants for the volumes of plasma, extracellular fluid, and tissue. We explored this model using a data set of 553 drugs for which VDss and plasma protein binding were available in humans. Eighteen percent of cases (102 compounds) did not obey the Øie-Tozer model, with the rearranged equation giving an aberrant fut value (fut < 0 or fut > 1), in particular for compounds with VDss < 0.6 l/kg and fup > 0.1. Further analysis of this group of compounds revealed patterns in physicochemical attributes with a high proportion exemplified by logP less than 0 (i.e., very hydrophilic), polar surface area >150 Å2, and a difference between logP and logD >2.5. In addition there was a high representation of certain drug classes including anti-infectives as well as neuromuscular blockers and contrast agents. The majority of compounds were also found to have literature evidence, implicating active transport processes in their disposition. This analysis provides some important insights for pharmacokinetic optimization in this particular chemical space, as well as in the application of the Øie-Tozer model for predicting volume of distribution in humans.
Volume of distribution is a key pharmacokinetic parameter relating the systemic concentration of drug to the amount in the body. It is generally considered to be a theoretical rather than a physical term, which can be expressed in various forms including VD central, VD at steady-state, and VD terminal (Gibaldi et al., 1969). VD central (VDc) represents the initial dilution volume of the drug, calculated as dose divided by the initial plasma concentration (C0 extrapolated) after an intravenous dose and is usually small as equilibration into tissues has yet to occur. VD terminal (VDz or VDβ) is calculated as clearance divided by the terminal phase rate constant and represents the stage when distribution is complete with redistribution from tissues to plasma being predominant. As such it is heavily dependent on the terminal phase rate constant, and characterization of this phase can prove problematic as the limits of bioanalytical quantification are reached. VD steady state (VDss) can be thought of as a “time-averaged” volume lying somewhere between VDc and VDz, and when tissue concentrations have reached a maximum. This VD parameter is calculated as the product of dose and area under the first moment curve divided by the square of the area under the curve and is generally considered most useful in assessing potential dosing regimens and expected accumulation in multiple dosing scenarios.
The determinants of volume of distribution tend to include tissue affinity driven by lipophilic and electrostatic interactions with membrane phospholipids as well as pH partition mechanisms into organelles such as lysosomes (Smith, 1997; Van de Waterbeemd et al., 2001; Lombardo et al., 2002, 2004; Obach et al., 2008). Plasma protein binding is also important, and it is generally driven by lipophilicity plus anionic characteristics due to an electrostatic interaction with a basic residue in the most abundant plasma protein, albumin. Another contributing factor that has received much attention recently is the role of active transport processes in the volume of distribution (Grover and Benet, 2009; Shugarts and Benet, 2009). By analyzing literature data on pharmacokinetic interactions at the transporter level, Grover and Benet (2009) were able to show that the greatest impact of transporters on VD was once distribution equilibrium had occurred, i.e., VDss and VDz. In addition, uptake and efflux interactions at the liver generally decreased VD, whereas efflux interactions at the kidney generally increased VD.
Transport of a xenobiotic against its concentration gradient, using ATP hydrolysis or facilitated by an opposing endogenous concentration gradient, is an important process in drug disposition. The proteins responsible are expressed in many tissues including but not limited to intestine, liver, kidney, and brain. This topic has become, over the years, an area of major focus and a multitude of active transporters implicated in drug transport have been identified, cloned, and recombinantly expressed (Xia et al., 2008). They typically fall into two categories: uptake (from luminal/vascular to tissue), e.g., organic anion-transporting polypeptide, and efflux (from tissue to luminal/vascular), e.g., multidrug resistance protein 1 (MDR1) or P-glycoprotein. The most well known and widely studied transporter is P-glycoprotein (MDR1) identified as playing a key role in limiting oral absorption and central nervous system penetration as well as mediating biliary excretion of substrates, all as a result of high expression at the gut wall, blood-brain barrier, and hepatocyte sinusoidal/bile canaliculi membranes, respectively.
Øie and Tozer (1979) proposed the model for volume of distribution at steady state described by plasma and tissue drug binding, building on the work of Gillette (1976) by including a term for the extravascular/intravascular ratio of nonspecific drug-binding sites or amount of binding protein. The primary model assumption is that steady state is reached via purely passive diffusion phenomena and does not account for active transport of drug against concentration gradients.
More recently, this model was shown to be useful in predicting human VDss from animal data by rearrangement of the equation to describe a tissue free fraction. Tissue binding is generally considered to be consistent across species as this is typically driven by hydrophobic and electrostatic interactions with common cellular constituents such as membrane phospholipids. By using VDss and plasma-free fraction in preclinical species to generate a “species-independent” tissue free fraction, this figure could be used together with a human plasma free fraction in the original form of the model to calculate human VDss (Lombardo et al., 2002, 2004). The aim of the present work was to explore the Øie-Tozer model further using a data set of VDss and plasma protein binding values in human on 553 compounds. A reasonable proportion of the data set (18%) did not obey the model, giving rise to aberrant fut values either less than 0 or greater than 1. The sensitivity of the model to the exact value of extravascular/intravascular binding protein ratio was also demonstrated. Moreover, on closer analysis, the violating compounds revealed trends in physicochemical properties and therapeutic class, with the majority of them having literature data supporting their action as substrates of various active transporters. Finally, we show that for these types of compounds, the application of the Øie-Tozer equation could lead to erroneous predictions if aberrant fut values are not observed in animals and then used for human VDss prediction.
Materials and Methods
This analysis used a database of human intravenous pharmacokinetic parameters recently published (Obach et al., 2008). Within this 670-compound data set, there were 553 compounds for which human plasma protein-binding data were available. Data on these 553 compounds formed the basis for this work.
The physiological model for VDss as defined by Øie and Tozer (1979) is as follows:
 where fup is the fraction unbound in plasma, fut is the fraction unbound in tissues, and RE/I is the extravascular/intravascular ratio of binding proteins (usually 1.4 for albumin). VP, VE, and VR refer to the volumes of plasma, extracellular fluid, and remainder fluid with values of 0.0436, 0.151, and 0.38 l/kg, respectively, in humans. This equation was rearranged to express fut in terms of VDss and fup as follows:
where fup is the fraction unbound in plasma, fut is the fraction unbound in tissues, and RE/I is the extravascular/intravascular ratio of binding proteins (usually 1.4 for albumin). VP, VE, and VR refer to the volumes of plasma, extracellular fluid, and remainder fluid with values of 0.0436, 0.151, and 0.38 l/kg, respectively, in humans. This equation was rearranged to express fut in terms of VDss and fup as follows:
 The sensitivity of the RE/I index on generating an aberrant fut value for 102 compounds was assessed by varying this parameter from 0.1 to 2.5 in 0.1-unit increments with all volume terms (VP, VE, and VR) held constant.
The sensitivity of the RE/I index on generating an aberrant fut value for 102 compounds was assessed by varying this parameter from 0.1 to 2.5 in 0.1-unit increments with all volume terms (VP, VE, and VR) held constant.
Physicochemical descriptors were calculated using VolSurf+ (version 1.0; Molecular Discovery Ltd., Pinner, Middlesex, UK) and included lipophilicity parameters (logP and logD7.4) as well as polar surface area (PSA) and molecular weight. Literature searches for compound-related information pertaining to substrates of active transport processes were undertaken using SciFinder 2007.
Results
The rearranged Øie-Tozer equation expressing fut in terms of VDss and fup gave aberrant results for 102 of 553 compounds, where fut was less than 0 or greater than 1. The 102 outlier compounds all had VDss of less than 0.6 l/kg and as illustrated in Fig. 1 fall into two classes with respect to their plasma protein binding; 12 compounds were highly plasma protein bound with fup < 0.1 and VDss < 0.2 l/kg, whereas the remaining 90 compounds had fup > 0.1 with a large proportion showing no binding to plasma proteins (fup = 1).

Plot of volume of distribution at steady state against log plasma free fraction for the 102 compounds, which gave rise to an aberrant tissue free fraction using the rearranged Øie-Tozer equation. Plasma free fraction less than 0.1 (○) and greater than 0.1 (●).
Based on this finding, simulations were performed across a range of VDss and fup values as shown in Fig. 2. From this analysis it is clear that for high VDss (1 and 3 l/kg) there are no model violation occurrences across the range of fup. However, in the two low VDss situations (0.3 and 0.5 l/kg) there are two separate threshold values of fup, which lead to aberrant fut values (fup > 0.3 and > 0.85, respectively). In the case of very low VDss (0.1 l/kg), generally accepted as the distributional volume of albumin, the model fails across the entire range of fup.
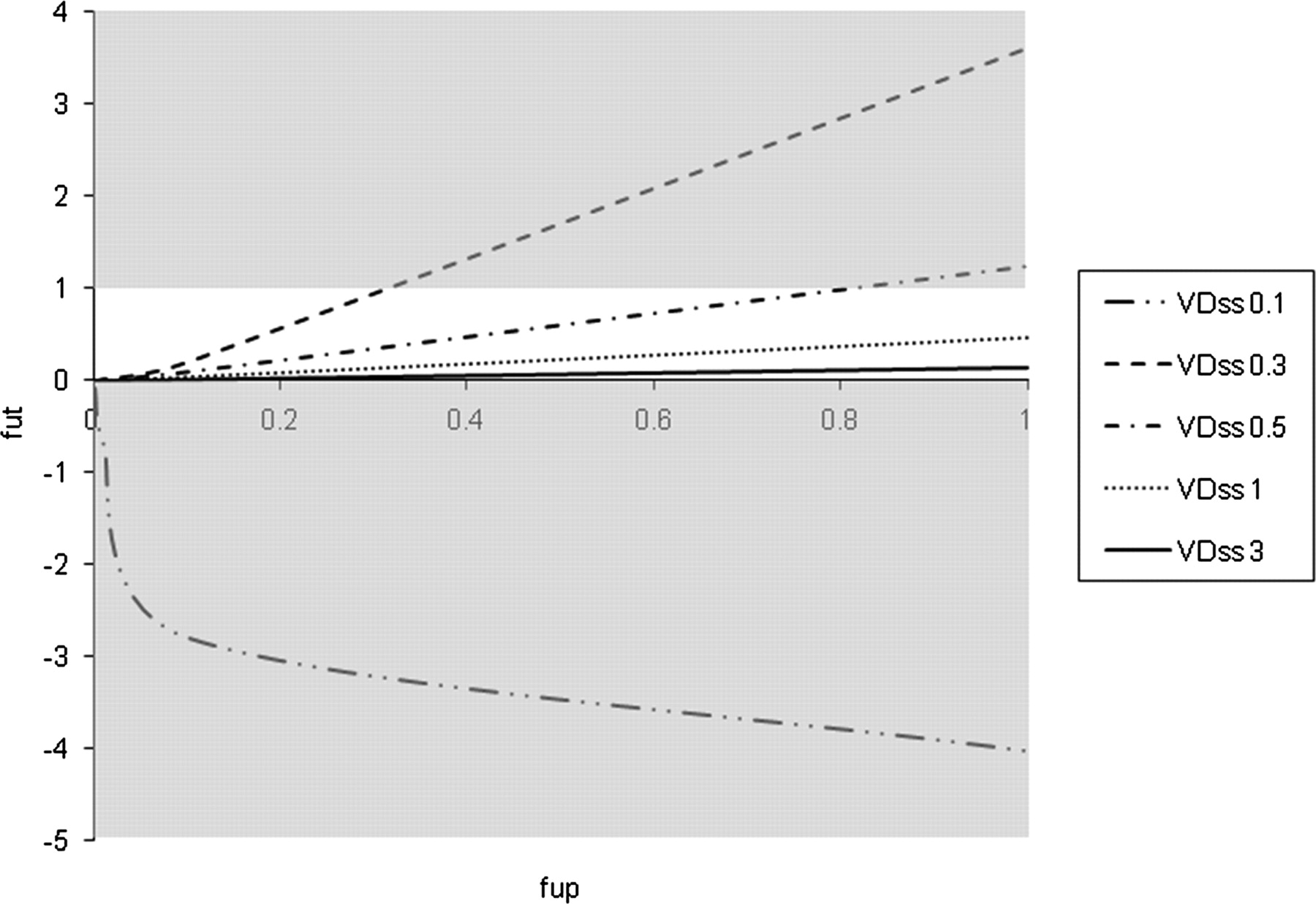
Simulations of the Øie-Tozer model for hypothetical compounds with VDss of 0.1, 0.3, 0.5, 1, and 3 l/kg with varying fup, illustrating model violations and the scenarios where fut becomes aberrant (gray shading).
In light of the number of model violations (102 of 553) the sensitivity of the extravascular/intravascular ratio of drug binding protein (RE/I) parameter was explored. Typically this is set at 1.4 based on the distribution of albumin. For the 102 outlier compounds, simulations were performed, varying RE/I from 0.1 (very low extravascular distribution of plasma proteins) to 2.5 (extremely high extravascular distribution of plasma proteins) with two separate classes based on extent of plasma protein binding (Fig. 3). Incremental increases in RE/I from 1.4 to 2.5 had no impact for either group on the proportion of compounds, generating fut values between 0 and 1. However, a change was observed when RE/I was lowered, with low fup (<0.1) compounds showing a much higher sensitivity to changes in this parameter. For example, if RE/I is lowered to unity, a substantial number of highly bound compounds (40%) generate fut values that fall within the acceptable range, whereas there is a negligible change for the low binding compounds.

Sensitivity analysis of the plasma protein RE/I for compounds with high (fup > 0.1, ♦) and low (fup < 0.1, □) plasma free fraction. All compound-dependent variables and physiological terms remained constant, while RE/I was varied from 0.1 to 2.5.
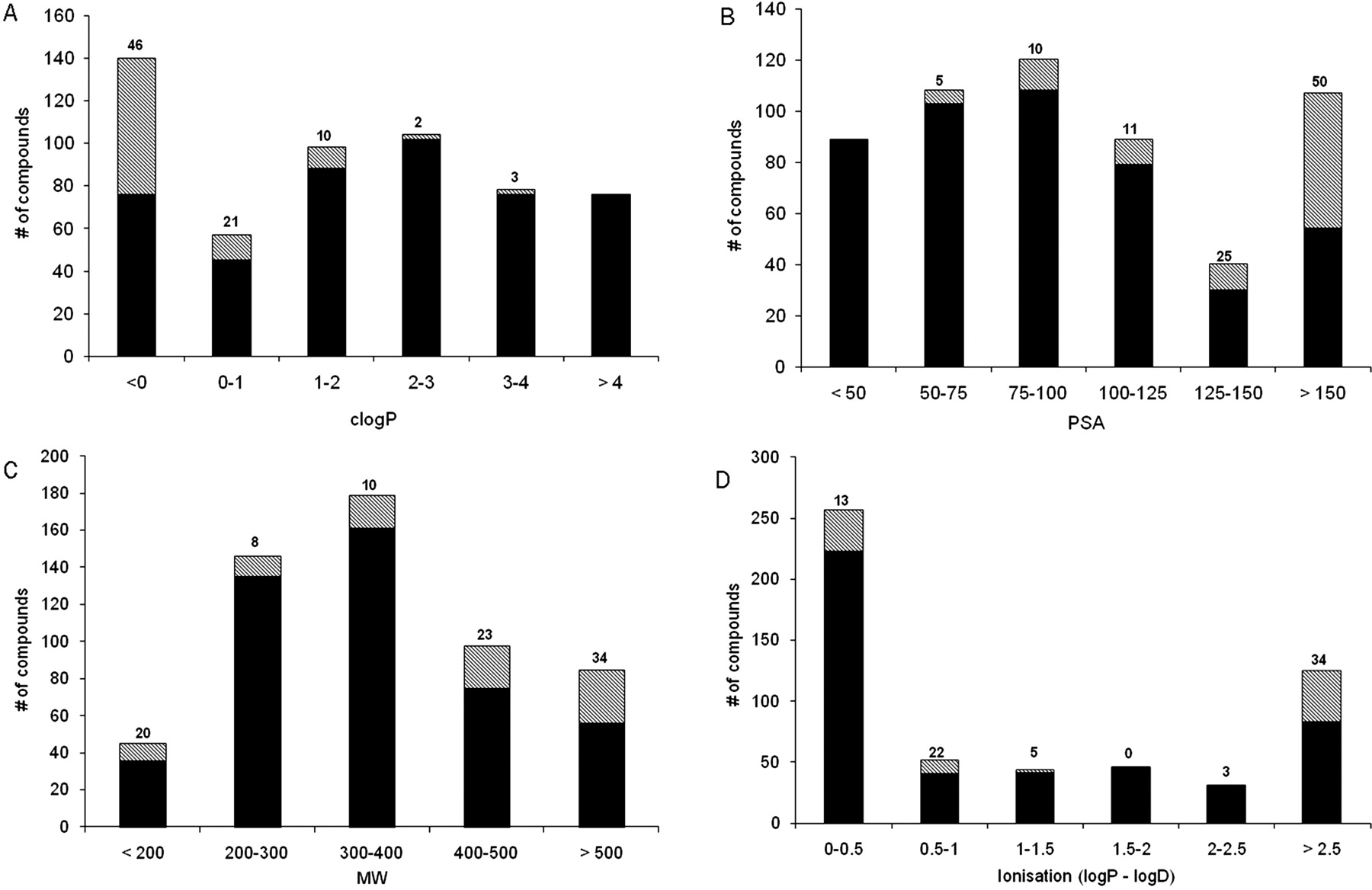
The physicochemical properties of the 90 outlier compounds, where VDss is less than 0.6 l/kg and fup is greater than 0.1, were investigated and are summarized in Fig. 4. Trends were observed with lipophilicity expressed as clogP, PSA, molecular weight, and extent of ionization (expressed as the difference between clogP and clogD) relative to the whole data set of 553 compounds. From these plots it is clear the majority of the aberrant compounds have very low lipophilicity with clogP typically less than 0; 46% of the compounds with clogP less than 0 give rise to an aberrant fut. High polarity is also an important contributing factor with 50% of the compounds with PSA >150 Å2 also leading to Øie-Tozer model violations. In addition, there is a trend with the degree of ionization where 34% of highly ionized compounds (defined as a difference between logP and logD greater than 2.5 log units) produce aberrant fut values. The trend with molecular weight is less clear although there are a greater proportion of outlier compounds with high molecular weight in excess of 400.

Trends in physicochemical properties observed between compounds with fut within normal range (0 < fut < 1; black) and compounds with aberrant fut (● data points in Fig. 1; i.e., 0 > fut or fut >1; hashed). A, clogP. B, polar surface area (in square angstrom). C, molecular weight. D, extent of ionization described by the difference between clogP and clogD at pH 7. Numbers above each bar represent percentage of compounds with aberrant fut within each range.
Possible explanations for the behavior of the 90 outlier compounds with low VDss and high fup were investigated further. Table 1 shows that a considerable number of these compounds have been shown to be substrates for various human active transport proteins.
Compounds that do not obey the Øie-Tozer model together with designated therapeutic activity and literature evidence for interaction with active transporters
Furthermore, to determine the predictive accuracy of the approach on the 102 violating compounds, calculations of human VDss were made using measured fup and three hypothetical fut values within the normal range (0.1, 0.5, and 0.9), as might normally be obtained from the rearranged Øie-Tozer equation with preclinical data. The summary statistics are displayed in Table 2.
Summary statistics on the performance of the Øie-Tozer model on the 102 outlier compounds applying various hypothetical estimates of fut (0.1, 0.5, and 0.9) as may usually be obtained from preclinical species
Discussion
The analysis presented has demonstrated a number of outlying scenarios for the Øie-Tozer model when VDss is low (<0.6 l/kg). Twelve of the compounds giving rise to aberrant fut values have fup < 0.1, probably because of variation at the upper end of the plasma protein-binding range, where accurate experimental determination of very low free fractions can become analytically challenging. In addition, the model is not applicable for compounds with VDss of 0.1 l/kg or lower: as fup tends to 0, VDss approximates to 0.105 l/kg. There is some sensitivity to the exact value of RE/I used for highly plasma-bound compounds as might be expected; altering the extravascular/intravascular distributional ratio of binding protein has a more significant impact on the fut because the distribution of these compounds is primarily driven by affinity for plasma proteins such as albumin. In cases of high plasma protein binding, an RE/I value of 1.4 may not be appropriate, and so the model should be applied with caution. Alternatively, information on the particular plasma proteins involved in drug binding may allow the RE/I to be tailored to compound-specific predictive application.
The remaining 90 compounds were explored further with respect to physicochemical attributes and possible pharmacokinetic explanations for the model violations. The Øie-Tozer model has a number of assumptions, which can aid understanding in cases in which aberrant fut values are obtained. The assumptions are 1) drug distribution is driven by nonspecific binding between blood and tissue, 2) rapid equilibration occurs between blood and tissue, 3) drug distributes uniformly within each organ or tissue, 4) there is no contribution from active transport processes, and 5) distributional processes are nonsaturating. In this regard, we have shown a substantial number of the 90 violating compounds have literature evidence supporting their action as substrates of human active transport proteins (Table 1). The transporters cited are typically of an efflux nature, limiting distribution into those tissues expressing the transport protein and consequently contributing toward limiting VDss to the volume of extracellular fluid (0.6 l/kg) or lower. This is not to say that all active transport substrates violate the Øie-Tozer model, e.g., the HIV protease inhibitors and angiotensin receptor antagonists are known to be actively transported and obey the Øie-Tozer model. In addition, it is not the case that low VDss is the sole driver for failures of the equation. Of the 216 compounds with VDss < 0.6 l/kg, 114 obey the model, whereas 102 compounds do not. Furthermore, it is not possible to substantiate the involvement of active transport as the exclusive determinant for whether or not model violations will be observed. There are obviously many other considerations including affinity for the transport protein, the magnitude of local, unbound concentrations, and dose size. These complicating factors make it difficult to assess the sensitivity of the model in this respect. For example, some compounds may give rise to back-calculated fut values that are realistic (i.e., between 0 and 1) but which remain inaccurate because of one or more model assumptions; e.g., many of the statins (organic anion-transporting polypeptide 1B1 substrates) are low VDss, high fup compounds, which in principle obey the model. Nevertheless, in cases of low VDss and high plasma free fraction, this analysis implicates the involvement of active transporters in limiting tissue partitioning.
It has been observed that tissue/plasma partition ratios are elevated in certain tissues in mdr1a knockout mice relative to wild-type mice, albeit not for every compound studied and in all tissues (Lee et al., 2009). An analysis of published reports demonstrating changes in distribution volume, in animals and humans, mediated by drug-drug interactions, genetic polymorphism, or gene knockout, showed some similar trends; uptake interactions at the liver tended to cause a decrease in VDss. However, the efflux interactions at the liver did not trend in the opposing direction, which could be a consequence of assessing interactions in a tissue generally considered part of the central compartment (Grover and Benet, 2009). This work highlights the potential complexity of drug distribution; a paradigm shift from simple passive diffusion phenomena to more intricate, active mechanisms including efflux, uptake, and intracellular sequestration. In addition, Dobson and Kell (2008) concluded that active transport processes are fundamental in determining drug disposition and are probably more common than usually assumed, citing the mounting evidence in the literature on specific drug examples as well as observations that drugs can concentrate in specific tissues.
It is clear that a large proportion of the 90 compounds are β-lactam antibiotics. This class of compounds was intentionally excluded from the rule-of-5 analysis because these natural products (and close derivatives) are highly likely to have “evolved” transport protein interactions (Lipinski et al., 1997; Ganesan, 2008). In addition, there are examples in the literature of this class of compounds accumulating in specific tissues by various mechanisms, a further model assumption violation. The β-lactam antibiotics have been shown to accumulate in the choroid plexus of rat via an active carrier-mediated transport process (Nohjoh et al., 1989). The aminoglycosides listed in Table 1, including amikacin, gentamicin, isepamicin, kanamycin, netilmicin, and tobramycin, have been shown to selectively accumulate in renal cortex, leading to renal injury. This selective accumulation is thought to be mediated by megalin, a large endocytic receptor abundantly expressed in renal proximal tubules (Tod et al., 2000; Nagai, 2006). Furthermore, a similar mechanism by which these compounds accumulate in sensory hair cells, leading to ototoxicity, has also been identified (Hashino et al., 1997). After receptor-mediated endocytosis, the compounds are transported by vesicular trafficking into lysosomes. Accumulation of drug leads to lysosomal disruption and rupture, with subsequent hair cell degeneration. Compare this active mechanism of lysosomal and mitochondrial trapping to that generally regarded for basic drugs for which a passive, pH partition mechanism is implicated (Okumura et al., 1989; Daniel and Wojcikowski, 1997). Contrast agents are also well represented, including a number of iodinated compounds such as iohexol as well as the gadolinium-containing compound, gadoversetamide. These agents are typically limited to the intravascular and extracellular fluid spaces and are renally cleared by glomerular filtration, in line with their intended clinical imaging applications. However, cytochemistry studies with the diagnostic indicator dye, fluorescein, have demonstrated active transport-driven renal accumulation in mitochondria (Masereeuw et al., 1994).
In addition to model assumptions concerning no active transport processes and uniform tissue distribution, there is also a requirement that distributional characteristics are nonsaturating. This could explain cases such as cefazolin for which plasma protein binding has been shown to be nonlinear in rat (Tsuji et al., 1983). The free fraction in rat serum at 10, 100, and 200 μg/ml was measured as 11, 20, and 41%, respectively. This marked change in plasma protein binding would probably shift the distributional behavior in tissues.
The Øie-Tozer model has been effectively applied to the prediction of human VDss for basic and neutral compounds (Obach et al., 1997; Lombardo et al., 2002, 2004). In this approach, the rearranged Øie-Tozer equation is used together with VDss and fup in preclinical species to calculate fut. This value is considered species-independent because tissue binding tends to be determined by the extent of interaction with phospholipid membranes. This fut together with experimental human fup measurements can be put into the standard form of the Øie-Tozer model to generate VDss for human (Obach et al., 1997). To determine the predictive accuracy of the approach on the 102 violating compounds, calculations of VDss were made using measured fup and three hypothetical fut values within the normal range (0.1, 0.5, and 0.9), as might normally be obtained from the rearranged Øie-Tozer equation with preclinical data. From the summary statistics displayed in Table 2, it is clear that when apparently normal fut values from preclinical species are used, large errors in prediction can be observed; assuming passive diffusion-mediated distribution leads, in the vast majority of cases, to overprediction of human VDss. Even when the more likely scenario of low tissue binding is applied (fut 0.9), the percentage of predictions with less than 2-fold error is lower than 60%, with a maximum error of 9-fold in this test set. The errors observed could be further exacerbated by the high proportion of actively transported drugs within the 102-compound set, especially given the known species differences in transporter expression and activity.
Multiple linear regression approaches have also been applied to the calculation of fut using two experimentally determined physicochemical properties, ElogD and fraction ionized at pH 7.4 (Lombardo et al., 2002, 2004). With the exception of the borderline example, metronidazole, for which a slightly different plasma free fraction was reported, there are no violating compounds present in these original reports. However, in recent work in which the ElogD approach was modified to a high-performance liquid chromatography-immobilized artificial membrane measurement, a number of acidic compounds were used, and, despite some of them giving rise to aberrant fut values (log fut > 0), they were included in the subsequent model building (Sui et al., 2009). The present investigation highlights the importance of careful application of the Øie-Tozer model and the need to be aware of the potential for aberrant fut values. In the same manner, and as shown in this work, the generation of an aberrant fut parameter from preclinical data, although not an all-encompassing diagnostic, can give some useful insights into potential disposition properties of novel chemical entities, implying active transport, selective tissue accumulation, or nonlinear, nonuniform distributional behavior. The judicious application of the Øie-Tozer model to predictions of human VDss for novel compounds is also noteworthy, in cases in which the physicochemical property profile or drug class overlaps with that demonstrated in this analysis. Further work in the field of drug transporters will help elucidate the nature of drug distributional behavior and provide further insights to aid drug design.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032458.
-
ABBREVIATIONS:
- VD
- volume of distribution
- VDss
- volume of distribution at steady-state
- Mdr/mdr
- multidrug resistance protein
- fut
- fraction unbound in tissue
- fup
- fraction unbound in plasma
- RE/I
- extravascular-to-intravascular binding protein ratio
- PSA
- polar surface area.
- Received January 27, 2010.
- Accepted April 7, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}