


Abstract
Although reversible CYP3A inhibition testing is well established for predicting the drug-drug interaction potential of clinical candidates, time-dependent inhibition (TDI) has become the focus of drug designers only recently. Failure of several late-stage clinical candidates has been attributed to TDI, and this mechanism is also suspected to play a role in liver toxicities often observed in preclinical species. Measurement of enzyme inactivation rates (kinact and KI) is technically challenging, and a great deal of variability can be found in the literature. In this article, we have evaluated the TDI potential for 400 registered drugs using a high-throughput assay format based on determination of the inactivation rate (kobs) at a single concentration of test compound (10 μM). The advantages of this new assay format are highlighted by comparison with data generated using the IC50 shift assay, a current standard approach for preliminary assessment of TDI. With use of an empirically defined positive/negative kobs bin of 0.02 min−1, only 4% of registered drugs were found to be positive. This proportion increased to more than 20% when in-house lead optimization molecules were considered, emphasizing the importance of identifying this property in selection of promising drug candidates. Finally, it is suggested that the data and technology described here may be a good basis for building structure-activity relationships and in silico modeling.
Introduction
Cytochromes P450 (P450s) are major metabolizing enzymes involved in the clearance of a large number of drugs (Liu et al., 2007). Inhibition of P450 enzymes by coadministered drugs can lead to overexposure and has been attributed to the withdrawal of several drugs from the market, of which mibefradil is one example (Krayenbuhl et al., 1999). A major P450 isoform family, CYP3A is particularly important because it is involved in the metabolism of numerous marketed drugs (Rendic, 2002). For new chemical entities (NCEs), inhibition of CYP3A is a major drawback. Risk evaluation for this particular aspect has been routine in industry since the early 1990s with in vitro assays based on recombinant CYP3A and fluorogenic substrates (Crespi et al., 1997). These assays have evolved to use of P450 isoform-specific drug substrates and more relevant in vitro drug metabolism systems, e.g., midazolam 1′-hydroxylation in human liver microsomes, enabling more reliable in vitro-in vivo extrapolation of results (Foti et al., 2010). An additional complexity for evaluating the drug interaction potential of a CYP3A inhibitor resides in the mechanism of inhibition. Time-dependent inhibition (TDI) has been described for numerous drugs (Zhou et al., 2005) and is characterized by an increase in inhibitory potency with compound turnover. Potential mechanisms for this include the formation of a tight-binding, quasi-irreversible inhibitory metabolite complex or the inactivation of P450 enzymes by covalent adduct formation (Murray, 2009; VandenBrink and Isoherranen, 2010).
TDI often results in more severe drug interactions as restoration of activity generally requires de novo synthesis of the enzyme (Kalgutkar et al., 2007). In addition, in some cases, protein adducts, formed via TDI, may lead to autoimmune reactions and severe liver toxicities (Pirmohamed et al., 1996). Nefazodone, for which as yet unexplained and rare liver toxicities led to the market withdrawal, may be such an example (http://www.rxlist.com). Having robust assays in place to detect research compounds with these potential liabilities at early stages in drug discovery is extremely important.
The evaluation of time-dependent inhibition is one of the most challenging tasks for P450 enzymologists, requiring complex experimental design, a large number of single incubations per compound, and complex data analysis. This complexity has been recently reviewed, and conceptual errors that may explain the large variation in published results have been described previously (Ghanbari et al., 2006; Yang et al., 2007). The characteristic parameters for TDI, kinact, the maximal inactivation rate, and KI, the concentration at half kinact, and their ratio kinact/KI are typically used for risk evaluation. Results of attempts to use excerpts of these experiments to screen a large number of compounds have been published (Atkinson et al., 2005; Lim et al., 2005; Obach et al., 2007; Perloff et al., 2009). The IC50 shift method, which is essentially an extension of the classic high-throughput testing for reversible inhibition, is the most popular because of its easy setup. The relationship between the shifted IC50 or fold shift in IC50 to kinact or KI requires careful evaluation and is difficult to use for TDI risk assessment (Krippendorff et al., 2009; Burt et al., 2010). However, Obach et al., 2007 have recently proposed a risk ranking model based on the shifted IC50. As an alternative, single concentration, single time point assays were reported for which percentage activity remaining or apparent partition ratio was used for risk evaluation (Lim et al., 2005; Watanabe et al., 2007).
In the present study, we assess a new TDI risk evaluation method based on the measurement of the observed first-order enzyme inactivation rate constant, kobs, at a relatively high test compound concentration of 10 μM. This method includes the crucial high dilution step important for TDI studies. The kinetic parameters kinact and KI were measured for 63 known drugs and compared with literature values. Inactivation rates at 10 μM were generated for a large set of 400 marketed reference compounds and rank-ordered for risk evaluation.
Materials and Methods
Reagents and Chemicals.
Midazolam was purchased from Sequoia Research Products Ltd. (Pangbourne, UK), and 1′-hydroxymidazolam-D4 was purchased from Cerilliant Corporation (Round Rock, TX). Drugs and Novartis proprietary compounds were obtained from the Novartis compound store as 10 mM stock solutions in dimethyl sulfoxide (DMSO). Their purity and concentration were controlled using various techniques. NADPH, 1′-hydroxymidazolam, and all other reagents, chemicals, and buffer salts were purchased from Sigma-Aldrich (St. Louis, MO) or Fluka (Buchs, Switzerland). Water, formic acid, and acetonitrile for LC-MS analysis were purchased in analytical grade from Merck (Darmstadt, Germany).
Human Liver Microsomes.
Pools of human liver microsomes from 50 individual donors (lot 82087) were obtained from BD Biosciences (Woburn, MA). The microsomes were characterized by the vendor with regard to the levels of enzyme-selective marker activities (CYP1A2, phenacetin O-deethylation; CYP2A6, coumarin 7-hydroxylation; CYP2B6, (S)-mephenytoin N-demethylation; CYP2C8, paclitaxel 6α-hydroxylation; CYP2C9, diclofenac 4′-hydroxylation; CYP2C19, (S)-mephenytoin 4′-hydroxylation; CYP2D6, bufuralol 1′-hydroxylation; CYP2E1, chlorzoxazone 6-hydroxylation; CYP3A4, testosterone 6β-hydroxylation; CYP4A11, lauric acid 12-hydroxylation; and flavin-containing monooxygenase, methyl p-tolyl sulfide oxidation).
Time-Dependent Inhibition.
To determine kinact and KI, six test compound concentrations (from 2.5 to 50 μM final concentration) were obtained by dispensing 2.5 to 50 nl of 10 mM stock solutions using an Labcyte ECHO 520 acoustic dispenser (Bucher Biotec, Basel, Switzerland) into an empty 96-well plate shortly before the incubation was started by the addition of 10 μl of 0.5 mg/ml human liver microsomes and 1 mM NADPH in 50 mM sodium phosphate buffer (pH 7.5, prewarmed at 37°C) using a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Vantaa, Finland). The DMSO concentration was adjusted to 0.5% in all incubations. For screening experiments, only 10 nl of the stock solutions of test compounds were dispensed for a 10 μM final concentration and a DMSO content of 0.1%. The plates were then preincubated at 37°C in an incubator (ELMI Skyline DTS-4; LTF Labortechnik, Wasserburg, Germany) for 0, 10, 20, and 30 min, and the residual CYP3A activity was determined by the addition of 90 μl (10-fold dilution) of 10 μM midazolam, 0.06 μM 1′-hydroxymidazolam-D4 (internal standard), and 1 mM NADPH in 50 mM sodium phosphate buffer (pH 7.5, prewarmed at 37°C) using the Multidrop Combi Reagent Dispenser. Addition of the internal standard 1′-hydroxymidazolam-D4 to the incubation rather than to the stop solution was found to improve assay robustness. The plates were incubated for 8 additional min before being stopped by the addition of 1 volume (100 μl) of ice-cold acetonitrile. Stopped incubation plates were stored at −20°C overnight and centrifuged at 5000g for 35 min at 4°C to pellet precipitated material. Aliquots of the supernatants were analyzed for 1′-hydroxymidazolam and 1′-hydroxymidazolam-D4 by LC-MS. Under these conditions, the inactivation rate of 10 μM verapamil was measured 80 times (20 assays) over a period of 6 months and did not deviate significantly from the value of 0.073 min−1 (coefficient of variation = 8%, minimum = 0.060 min−1, and maximum = 0.086 min−1).
Reversible Inhibition.
Dilution ranges of test compounds were obtained by dispensing 2.5 to 100 nl of 10 mM test article stock solutions in DMSO directly into the wells of a 384-well microplate by acoustic dispensing (0.5, 1, 2, 3, 5, 10, 15, and 20 μM final concentrations). For the definition of enzyme activity range (0–100%), 16 wells were filled with 100 nl of DMSO only (100%) and 16 wells with 100 nl of 5 mM ketoconazole (0% activity, 5 μM final concentration). Ten nanoliters of 10 mM midazolam (10 mM in DMSO, 2 μM final concentration) and 5 nl of 1′-hydroxymidazolam-D4 (0.28 mM in DMSO, 0.028 μM final concentration) were added as substrate and internal standard into every well of the 384-well microplate. All wells were supplemented to 100 nl with pure DMSO using the ECHO 520 acoustic dispenser (0.2% final DMSO content). The incubation was started by the addition of 50 μl of a mix of 0.05 mg/ml human liver microsomes and 1 mM NADPH dissolved in 50 mM sodium phosphate buffer (pH 7.5, prewarmed at 37°C) using a Multidrop Combi Reagent Dispenser. The plate was immediately placed in an incubator (ELMI Skyline DTS-4) and incubated for 10 min before the reaction was stopped by the addition of 50 μl of ice-cold acetonitrile containing 1 μM alprenolol as an additional internal standard for LC-MS analysis.
LC-MS Analysis.
Analysis of samples was performed on a high-performance liquid chromatography-tandem mass spectrometry system consisting of a TSQ Quantum Discovery MAX mass spectrometer controlled by QuickQuan 2.0 and equipped with an electrospray ion source (Ion Max electrospray interface) from Thermo Fisher Scientific (Reinach, Switzerland), a CTC-HTS Pal autosampler (CTC Analytics, Zwingen, Switzerland) with a sample cooling unit (10°C), and a Rheos pump (model 2000; Thermo Fisher Scientific). Samples were separated on a Polar-RP column (2.1 × 50 mm, 3.5 μm; Phenomenex, Torrance, CA) protected by a guard column (2 × 4 mm) containing the same material (provided by Brechbühler AG, Schlieren, Switzerland) using an isocratic mobile phase of water-acetonitrile (65:35) containing 0.1% formic acid at a flow rate of 400 μl/min for 2 min. The injection volume was 20 μl, and the first 0.5 min of eluent were diverted to waste to protect the ion source from salts and polar impurities. 1′-Hydroxymidazolam and 1′-hydroxymidazolam-D4 were detected in positive ion mode by selective reaction monitoring using the mass transition of 342 to 324 and 346 to 328, respectively, at a collision energy of 20 eV.
Data Analysis.
For time-dependent experiments, CYP3A enzyme activity in human liver microsomes was determined using the turnover of midazolam to 1′-hydroxymidazolam calculated by the ratio of 1′-hydroxymidazolam to the internal standard 1′-hydroxymidazolam-D4. The ratios were transformed to percentage active enzyme remaining (initial activity E0 = 100%) and plotted over the preincubation time. The first-order inactivation rate constant kobs was calculated by XLfit 4.3.2 (ID Business Solutions Ltd., Guildford, Surrey, UK) using model 503 with a range of 80% (a) and a background value (b) of 20%. When inactivation was strong, these values allowed the best estimate of initial slopes without the need for manual exclusion of the 30-min data point. Individual kobs values for test articles were corrected by the kobs value of the control containing DMSO only. The percentage reversible inhibition was calculated by the ratio of 1′-hydroxymidazolam to 1′-hydroxymidazolam-D4 at preincubation time 0 min in relation to the ratio of the control containing DMSO only. If there was a strong reversible inhibition (percentage reversible inhibition >50%), no kobs values were calculated.
In full time-dependent inhibition experiments, the Michaelis-Menten relationship was used for the determination of the maximal inactivation rate constant kinact and the irreversible inhibition constant KI. The calculated kobs values were plotted over the inhibitor concentration, and kinact and KI were calculated by nonlinear regression using XLfit model 250.
The IC50 values were calculated by plotting the ratio of 1′-hydroxymidazolam to 1′-hydroxymidazolam-D4 against the inhibitor concentration in the incubation using XLfit model 205. Range and background were set to 100% and 0% using the DMSO control and the ketoconazole fully inhibited control.
Results
Experimental Design.
Evidence suggests that experimental design for the characterization of TDI is a major contributor to data variability in published results. Several authors devoted full articles with extensive data reviews in their discussion and proposed several important guidelines for TDI incubations (Ghanbari et al., 2006; Yang et al., 2007). In addition to following these guidelines, our method included the innovative use of an acoustic dispenser device. Noncontact dispensing of nanoliters of compound stock solutions in DMSO allows the use of very small incubation volumes (10 μl) while keeping DMSO concentrations to a minimum (0.1%). This low volume allows for a high dilution (1:10) of the preincubate and final stop in the same vessel of a standard 96-well microtiter plate. This simplified protocol minimizes errors introduced by successive sampling and dispensing steps used in most TDI protocols.
Experimental conditions were chosen to ascertain proper characterization of TDI and following the general rules described by Ghanbari et al. (2006): 1) the inactivator is diluted 10-fold after preincubation; 2) the length of the incubation time is short and the probe substrate concentration is high (approximately 5-fold Km); 3) the residual activity is corrected by a control with no inhibitor; and 4) initial slope values of residual activity versus time were used to derive the inactivation rate kobs. Because DMSO is known to have an impact on midazolam hydroxylation activity by CYP3A4 (Nishiya et al., 2010), it was maintained at a minimum (0.1%). For full kinetic studies, a range of eight concentrations of test compound up to 50 μM was used (including DMSO control) at four preincubation times (0, 5, 10, and 30 min). For some drugs, the range of concentrations was reduced to avoid excessive reversible inhibition during the incubation step.
From the time- and concentration-dependent loss of enzyme activity curves (Fig. 1A), an initial inactivation rate (kobs) was calculated which, when plotted against concentration (Fig. 1B), enabled the experimental determination of kinact and KI. For the time-dependent inhibition screen, an excerpt of the full kinetic studies, in which a single test compound concentration of 10 μM in the preincubation was tested, was chosen.
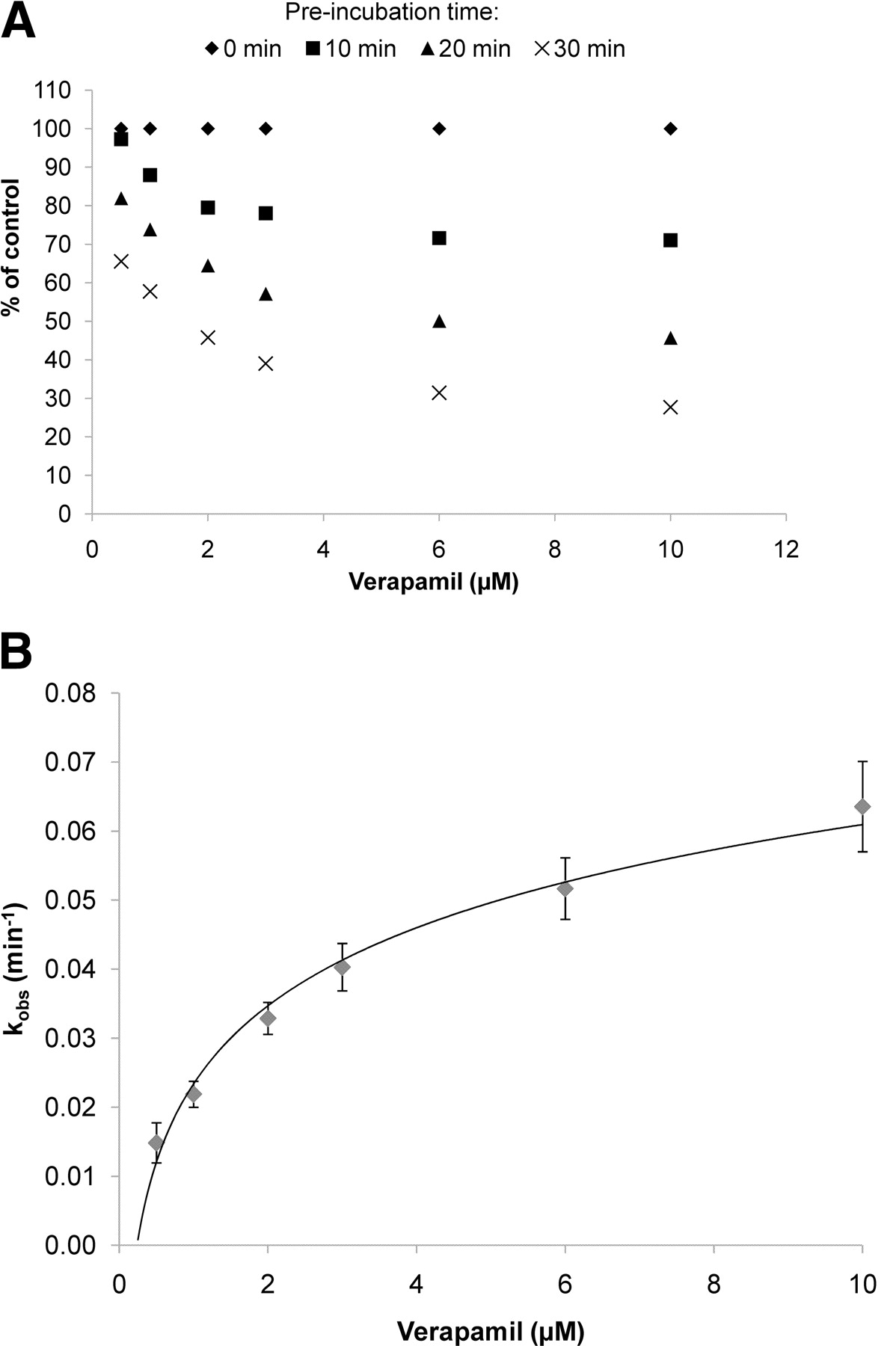
Typical graphical representation of data resulting from a full characterization of time-dependent inhibition. A, time- and concentration-dependent inactivation of CYP3A4-mediated midazolam hydroxylation by verapamil. B, relationship between the observed inactivation rate (kobs) and verapamil concentration.
Time- and Concentration-Dependent Inactivation.
Table 1 shows the comparison of kinetic parameters obtained for 21 drugs with data from literature references using similar conditions (pooled human liver microsomes and midazolam as the marker substrate for CYP3A). Although individual differences can be noted (nicardipine, raloxifene, and ticlopidine), the overall concordance is good, considering that there is already large variability among published values. For some compounds our experimental design did not allow us to measure KI and kinact because either the kinact (azamulin, nicardipine, and ritonavir) or the KI (azithromycin, isoniazid, and ThioTEPA) was too high. No TDI effect was observed for fluoxetine and paroxetine under the conditions used.
Kinetic parameters of CYP3A TDI in human liver microsomes and comparison with literature values
The TDI kinetic parameters measured for 63 drugs known or suspected to be time-dependent inhibitors are compiled in Table 2. The S.E.s given in the table are a measure of the goodness of fit and indicate which parameter is best described by the dataset (kinact or KI). Some drugs, although clearly time-dependent inactivators, were very potent reversible inhibitors, which precluded proper evaluation of their kinetic parameters for TDI (azamulin, mibefradil, nicardipine, and ritonavir). Khellin and isradipine were the only compounds for which kinact was clearly not reached at the highest concentration investigated (50 μM). For 16 additional drugs, we could not detect significant TDI in the concentration range tested, i.e., up to 50 μM (amiodarone, azithromycin, buspirone, carbamazepine, colforsin, diclofenac, duloxetine, eliprodil, fluoxetine, flutamide, isoniazid, lamotrigine, paroxetine, pulegone, ThioTEPA, and zileuton).
Kinetic parameters of CYP3A TDI (kinact and KI), risk factor ratio (kinact/KI), initial inactivation rate at 10 μM (kobs), IC50, and shifted IC50 for 63 drugs estimated after 10-fold dilution of the preincubate with 10 μM midazolam
Risk Evaluation.
The kinact/KI ratio is used throughout the literature (Obach et al., 2007) to evaluate TDI risk because this term combines both the intrinsic capacity of a compound to reduce enzyme activity (kinact) and the concentration at which this occurs (KI).
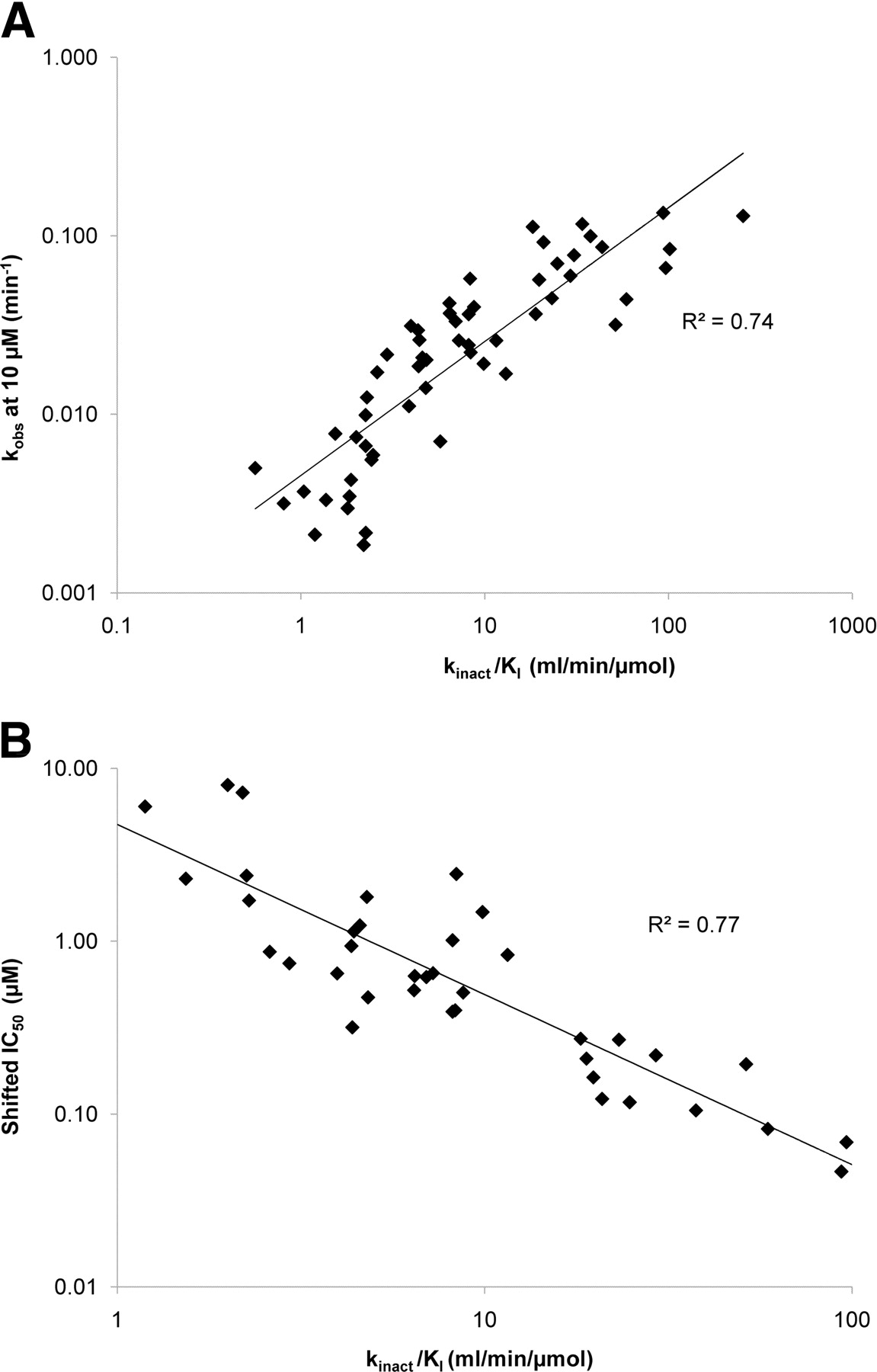
As described in previous studies (Obach et al., 2007), we found that the shifted IC50 correlates well (R2 = 0.77) with kinact/KI for the 40 drugs in our test set for which a numerical value for the shifted IC50 could be obtained (Fig. 2B; Table 2). The kobs at 10 μM correlated also nicely to the kinact/KI ratio (R2 = 0.74) (Fig. 2A; Table 2) with the additional advantage that a numerical value was available for significantly more compounds (57).

Relationship between time-dependent inhibitory potency and the risk factor ratio kinact/KI. A, data using kobs. B, data using shifted IC50. The solid lines are the lines of best fit. Values for kobs and shifted IC50 can be found in Table 2.
Time-Dependent Inhibition Screen and Compound Ranking.
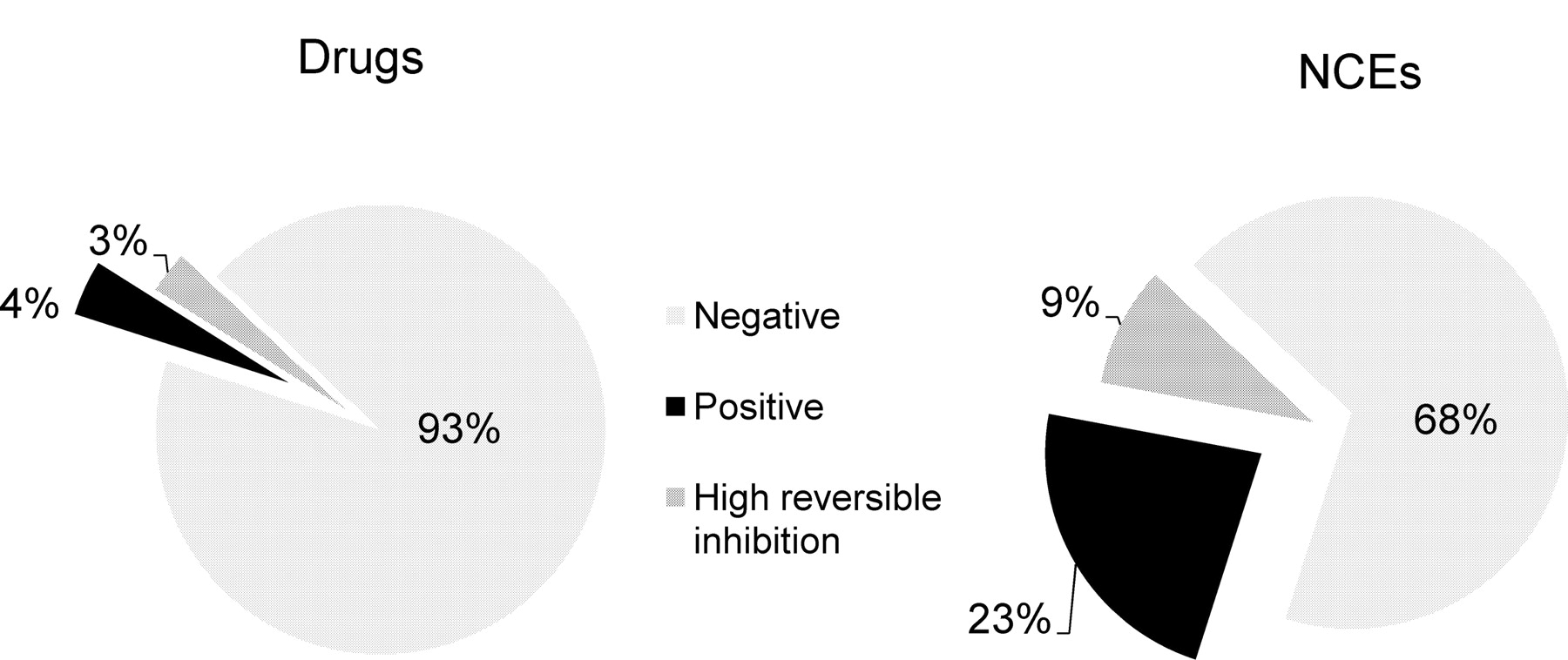
Figure 3 shows the inactivation rate (kobs) measured for 400 drugs (Supplemental Table S1) and approximately 4000 randomly selected NCEs from Novartis drug discovery programs. For World Drug Index drugs, the assay has been adapted to be able to measure kobs values higher than 0.1 min−1 with accuracy. The kobs value of the DMSO control was 0.004 min−1, i.e., at 37°C, 10% of the midazolam hydroxylase activity of human liver microsomes was spontaneously lost after 30 min of incubation. Of interest, the kobs values for test compounds increased continuously from values similar to the DMSO control to the high values for known potent time-dependent inhibitors such as troleandomycin. Only approximately 18% of the test compounds have a kobs similar to that of the DMSO control. From the shape of the curve in Fig. 3 and from positive drug benchmarks, a positive/negative bin border was set at a kobs value of 0.02 min−1. With use of this binning scheme, the percentage of TDI-positive drugs is very low at only 4% (Fig. 4). A similar proportion (3%) of drugs showed high reversible inhibition (percentage reversible inhibition >50%). The proportion of positive NCEs among a recent random set of approximately 4000 was much higher (23%). Those showing high reversible inhibition were also more abundant (9%).

Graphical representation of test compounds ranked by inactivation rate kobs.

Distribution of TDI-positive, TDI-negative, and strong reversible inhibitors among registered drugs and recent new chemical entities from Novartis.
Interplay with Reversible Inhibition.
The high and reversible inhibitory potency of human immunodeficiency protease inhibitors means that maximal TDI inactivation rates could not be reached without strong interference from reversible inhibition. However, as expected from the experimental design, all drugs and in-house compounds showing more than 50% competitive inhibition in this assay (3% of drugs and 9% of in-house compounds) were also shown to have IC50 values lower than 1 μM in the reversible inhibition assay when midazolam was at its Km concentration (Fig. 5). Of interest, however, up to 71% of TDI-positive NCEs were not flagged in the reversible inhibition assay (IC50 <1 μM, data not shown). Approximately 24% had no warning at all (IC50 >10 μM). This proportion increased to approximately 83 and 38% with registered drugs. Among TDI-negative drugs the same pattern can be seen. Only 2% of the TDI-negative drugs showed high reversible inhibition and only approximately 16% had IC50 values lower than 10 μM. Again, this proportion increased to 7 and 38% for NCEs.

Overlap of TDI ranking and reversible inhibition binning.
Discussion
The systematic and detailed investigation of TDI of P450 enzymes and its inclusion in drug interaction prediction algorithms in early drug development stages are relatively recent (Obach et al., 2007).
Kinetic parameters for a total of 63 drugs were measured to provide benchmarks for compound risk assessment (Table 2). When the TDI parameters measured with those published using similar test systems were compared, good concordance was found (Table 1). The variability seen between our data and literature values was similar to the variability seen between different literature references. All of the known strong positive time-dependent inhibitor compounds often used as benchmarks were also found to be positive in our test system (e.g., troleandomycin, verapamil, and erythromycin).
Several drugs had not been described as positive CYP3A4 time-dependent inhibitors to our knowledge: cilostamide, clemizole, dipyridamole, isradipine, motesanib, naftidrofuryl, nimodipine, omeprazole, oxatomide, ruboxistaurin, and tofisopam. Because most of these also show significant reversible inhibition of CYP3A4 (Table 2), it is likely that clinical relevance has been clarified by specific clinical drug interaction trials. Therapeutic concentrations of the calcium channel blocker isradipine were, for example, shown to have little effect on triazolam exposure in humans (Backman et al., 1999).
Full kinetic characterization of time-dependent inhibition necessitates a complex experimental setup and requires a laborious workflow, and numerous data points need to be collected and analyzed. When capacity is a limiting factor or to reduce the number of candidates for full characterization, a surrogate of the full experiment or a screening approach is useful. Obach et al. (2007) found a correlation between kinact/KI with the shifted IC50 obtained after preincubation of the test compound for 30 min. The composite parameter kinact/KI has been used and validated to estimate clinical drug interaction risk by these authors. We were able to reproduce this correlation with our method using a large set of drugs (Fig. 2A). In addition, we also found a good correlation between the inactivation rate at 10 μM (kobs) and kinact/KI (Fig. 2B). Disadvantages of using the IC50 shift assay for TDI risk assessment versus our method include the following: 1) a numerical value of the shifted IC50 is not always attainable without repeating the experiment and fine-tuning; 2) the magnitude of the shift may depend on the dilution factor; 3) strong reversible inhibition may interfere more dramatically and mask TDI; and 4) the presence of the substrate and its binding properties may influence the binding mode of the test compound and TDI mechanism. The two last arguments are particularly relevant in experiments in which dilution of the preincubate is smaller (2–5-fold) (Atkinson et al., 2005). In addition, measuring kobs was also less resource-intensive because it was possible to determine kobs accurately using only four preincubation time points, whereas proper IC50 determination requires six to eight concentrations spanning more than 2 orders of magnitude at least.
The choice of a concentration of 10 μM in the preincubation step was found to be a good compromise between solubility limitations of test compounds and their likelihood to reach this concentration level in human blood at therapeutic doses. The known exceptions are macrolide antibiotics. For example, erythromycin is a known time-dependent inhibitor with clinical relevance, for which steady-state therapeutic plasma concentrations reach values approaching KI (27 μM) and cause an increase in the AUC for midazolam after coadministration (Ito et al., 2003). However, most modern drugs are highly efficacious, and their therapeutic effects are seen at blood concentrations that rarely exceed micromolar levels.
CYP3A inactivation rates at 10 μM have been determined for 400 drugs (Supplemental Table S1) and approximately 4000 Novartis proprietary compounds. Most compounds showed some level of TDI with only approximately 18% behaving similarly to the DMSO control. The loss of enzyme activity for so many compounds suggests that it may be due to mechanisms other than reactive or tight binding of metabolites. Uncoupling of the catalytic cycle (futile cycling) induced by binding, turnover of compounds, and associated generation of reactive oxygen species may be an additional mechanism for TDI (Narasimhulu, 2007). Our method (based on kobs at 10 μM) correlates well with other methods for TDI risk assessment. Therefore, this method is a valid and robust approach for TDI risk assessment and is a reliable approach to prioritize research compounds for mechanistic kinact and KI determination.
We have chosen a binary categorization scheme with a kobs value of 0.020 min−1 as the lower limit for positive compounds. This threshold allows one to flag weaker time-dependent inhibitors such as clarithromycin as positive. Azithromycin is clearly negative with a kobs of 0.002 min−1. This finding is consistent with the published relative interaction potency in vivo (Ito et al., 2003). Examples of drugs with high KI, which are appropriately flagged as positive using this assay, include ethinylestradiol, tadalafil, and zafirlukast. Despite a kinact/KI ratio suggesting poor risk for erythromycin (Table 2), it is a clinically relevant time-dependent inhibitor because of its very high therapeutic systemic concentrations. In our assay, erythromycin had a kobs of 0.008 ± 0.003 min−1 (Supplemental Table S1) and is therefore considered to be TDI-negative. This example emphasizes that if high therapeutic concentrations are expected, the conditions of the assay described here should be adapted (higher preincubate concentration) or the bin border reduced accordingly.
The set of drugs used for this study was artificially enriched with all known time-dependent inhibitors. When an unbiased random drug set was tested, the proportion of TDI-positive compounds with a kobs value greater than 0.02 min−1 was only 4% (Fig. 3). In contrast, the proportion of positive NCEs among the 4000 preclinical candidates tested is much higher (23%). Among the NCEs found with kobs >0.02 min−1 (TDI-positive) more than 95% confirmed as positives in follow-up in vitro mechanistic studies (data not shown). The proportion of compounds showing high reversible inhibition also increased from 3 to 9%. Of interest, approximately 35% of the drugs and 24% of the NCEs that are TDI-positive showed no inhibition (IC50 >10 μM) in the standard assay for reversible inhibition, suggesting that TDI testing is required for full evaluation of the drug-drug interaction risk. Conversely, very few TDI-negative drugs (2%) and NCEs (7%) showed strong competitive CYP3A inhibition (IC50 <1 μM), suggesting that this TDI screen has a stronger filtering capacity than the reversible inhibition assay. More than half of the TDI-negative drugs with strong reversible inhibition were azole antifungals, which are well known for their potent CYP3A4 reversible inhibition. Despite strong reversible inhibition, itraconazole could be identified as a time-dependent inhibitor in a mechanistic study (Table 2). However, because itraconazole metabolites are known to also be strong inhibitors of CYP3A4 (Isoherranen et al., 2004), it cannot be excluded that TDI results from their formation and not from inactivation of the enzyme. TDI evaluation was not done systematically in earlier times of drug discovery; the low proportion of marketed drugs with TDI suggests that TDI-positive drugs were filtered out through their indirect effects or in late stage drug interaction clinical trials.
A rather poorly described aspect of TDI of CYP3A is the potential selectivity difference between CYP3A4 and CYP3A5. Midazolam hydroxylation is known to be catalyzed by both isoform and inhibitors have been shown to exhibit differential potencies on both isoforms (McConn et al., 2004). If significant turnover subsists in vivo because of a lower sensitivity toward CYP3A5, it may complicate considerably the prediction of CYP3A TDI in clinical outcomes. However, in preliminary experiments using human liver microsomes lacking the CYP3A5 isoform, we did not observe major differences in the TDI of benchmark inhibitors such as troleandomycin, nelfinavir, raloxifene, ruboxistaurin, tofisopam, or nefazodone. More experiments are needed to understand the affect of CYP3A5 on inactivation rates of midazolam hydroxylation.
Reversible inhibition has been shown to be substrate-dependent, and it is recommended that a second substrate, such as testosterone or nifedipine, be used in addition to midazolam. Because TDI parameters obtained with the dilution method do not include the competition aspect, they are likely to be independent of the substrate used (Watanabe et al., 2007).
Discrimination between TDI-positive drugs forming a metabolic intermediate complex versus those being authentic mechanism-based inactivators would be of great interest because the clinical risk is higher for the later (Lim et al., 2005; Kalgutkar et al., 2007). Combining the high-throughput screen described here with recently validated methods to distinguish a metabolic intermediate from mechanism-based inactivators seems to be possible and is currently under investigation.
The strategy proposed in this study has been shown to predict for kinact/KI ratios and offers some specific advantages over methods published previously. Risk evaluation and prioritization can take place at early discovery phases when structural modifications are still possible. Thanks to the large database of robust TDI data, modeling efforts to help chemists identify the metabolic spots likely to generate the inactivating molecular entity have been started. Isolation and characterization of these is virtually impossible because of their very nature (reactive intermediate); therefore, modeling with structure-property relationships seems to be the most appropriate approach for rational design of drugs with lower risk of irreversible inhibition.
Authorship Contributions
Participated in research design: Zimmerlin and Trunzer.
Conducted experiments: Zimmerlin and Trunzer.
Contributed new reagents or analytic tools: Zimmerlin and Trunzer.
Performed data analysis: Zimmerlin and Trunzer.
Wrote or contributed to the writing of the manuscript: Zimmerlin, Trunzer, and Faller.
Acknowledgments
We thank Rowan Stringer, Novartis Institutes for BioMedical Research, Horsham, UK, for careful reading of the manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.037911.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- P450
- cytochrome P450
- NCE
- new chemical entity
- TDI
- time-dependent inhibition
- DMSO
- dimethyl sulfoxide
- LC
- liquid chromatography
- MS
- mass spectrometry.

- Received December 23, 2010.
- Accepted February 25, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}