


Abstract
Selegiline was used as a model compound in a project aimed at comparing, evaluating, and integrating different in vitro approaches for the prediction of cytochrome P450 (P450)-catalyzed hepatic drug metabolism in humans (EUROCYP). Metabolic predictions were generated using homology modeling, cDNA-expressed P450 enzymes, human liver microsomes, primary cultured human hepatocytes, and precision-cut human liver slices. All of the in vitro systems correctly indicated the formation of two dealkylated metabolites, desmethylselegiline and methamphetamine. The metabolic instability of selegiline was demonstrated by all of the in vitro systems studied. Estimates of clearance varied from 16 l/h to 223 l/h. With the exception of one approach, all systems underpredicted the in vivo clearance in humans (236 l/h). Despite this, all approaches successfully classified selegiline as a high clearance compound. Homology modeling suggested the participation of CYP2B6 in the demethylation of selegiline and of CYP2D6 in the depropargylation of the drug. Studies with recombinant expressed enzymes and with human hepatic microsomal fraction supported the involvement of CYP2B6 but not of CYP2D6. These techniques also suggested the involvement of CYP1A2, CYP2C8, and CYP2C19 in the biotransformation of selegiline. In vitro, CYP2B6 was the most active form of P450 involved in selegiline metabolism. Metabolism by several enzymes operating in parallel implies a low interaction potential for the drug. None of the techniques alone was able to predict all aspects of the metabolic and kinetic behavior of selegiline in vivo. However, when used as an integrated package, all significant characteristics were predictable.
For more than a decade, in vitro techniques have had a well established role in the early phases of drug development in predicting the pharmacokinetic and metabolic behavior of new chemical entities. When properly used, predictions based on results from in vitro studies will save both development costs and time. In addition, they greatly reduce the number of experimental animals used by the industry in absorption, distribution, metabolism, and excretion studies. Support for the increased utilization of in vitro techniques in drug metabolism studies has come from regulatory authorities (CDER/FDA, 1997). Most of these techniques are easily applicable to high throughput screening, which has further promoted their use recently (Rodrigues, 1997).
Several in vitro methods for metabolic predictions are in common use, and even commercially available, but little attention has been paid to the validation of the systems in terms of quality of the predictions obtained and their usefulness in the process of drug development. In particular, comparisons of the data produced by different systems have been scarce. The present study was part of the EUROCYP project within the Biomed2 (Framework IV) program of the European Union. The goals of the project were to evaluate, compare, and integrate different in vitro approaches for the prediction of P450-catalyzed hepatic drug metabolism in humans for drug development. The final stage of the performance test was a “blind” evaluation of the metabolism of four model compounds using molecular modeling, hepatic subcellular fractions, recombinant expressed enzymes, hepatocytes and human liver slices. The results were compared with the existing human metabolic and pharmacokinetic data for each compound. One of the four model compounds was selegiline (SEL1). The others were almokalant (Andersson et al., 2001), carbamazepine (Pelkonen et al., 2001) and carvedilol (A. R. Boobis and O. Pelkonen, unpublished observation).
Selegiline is an irreversible inhibitor of the monoamine oxidase type B enzyme (Fowler et al., 1981). It is widely used alone or as an adjuvant to l-DOPA treatment in Parkinson's disease (The Parkinson Study Group, 1993; Koller, 1996; Myllylä et al., 1996). At the neuronal level, selegiline is also a dopamine reuptake inhibitor (Zsilla and Knoll, 1982; Tekes et al., 1988). More recently, interest has been directed toward the drug as a neuroprotective or neuronal rescue agent (Carrillo et al., 1991; Tatton and Greenwood, 1991; Mytileneou et al., 1997).
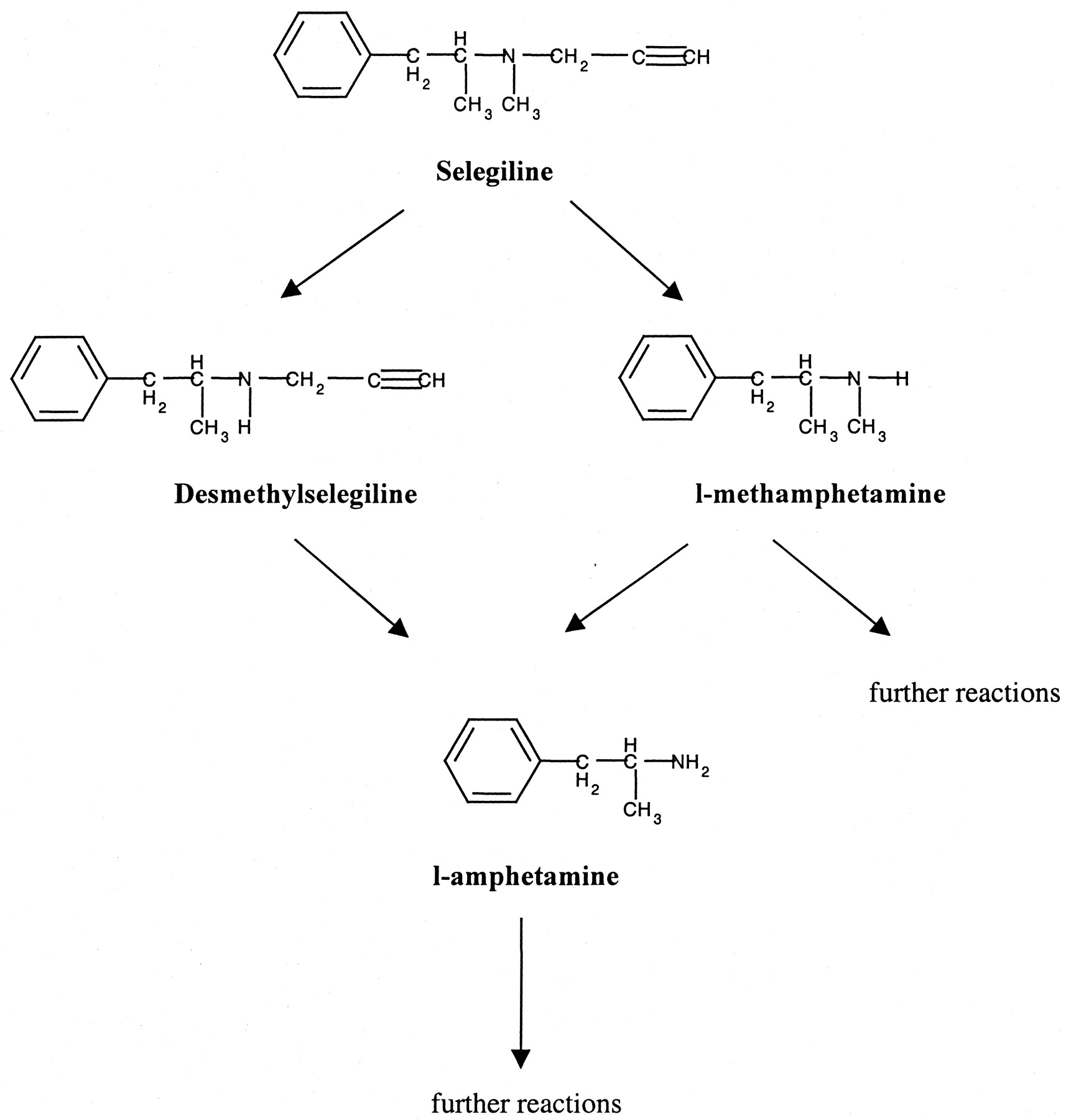
Selegiline is eliminated exclusively by biotransformation. Originally, methamphetamine (MA) and amphetamine (A) were identified as the major metabolic products in human urine and depropargylation with subsequent demethylation was postulated as a key metabolic pathway of the drug (Reynolds et al., 1978). Subsequently, a second major pathway, via desmethylselegiline (DMS), was reported (Yoshida et al., 1986). In addition, hydroxylated amphetamines have been reported in the urine of rats and humans dosed with selegiline, but these have been shown to be minor metabolites (Fig. 1) (Yoshida et al., 1986; Shin, 1997). Both initial dealkylations have been shown to be catalyzed by P450 enzymes (Yoshida et al., 1986). Recently, some work on the participating P450 enzymes has been published (Taavitsainen et al., 2000; Hidestrand et al., 2001), but no comparison of available candidate systems has yet been reported.

Metabolic pathways of selegiline in humans; only major products are shown.
Materials and Methods
The laboratories performing the incubations were blind to the identity of the compound at the time of the study, and the analytical laboratory was blind to the design of the study from which the samples being analyzed were derived, until after completion of that phase of the study. Because of blinding, incubation conditions could not be optimized for the particular compound under study which may have affected, especially, the clearance estimates. Since molecular modeling is based on the compound structure, modeling could not be blinded.
Materials. Selegiline hydrochloride [(-)-(R)-N,α-dimethyl-N-2-propynylphenethylamine hydrochloride] was obtained from Orion Pharma (Turku, Finland) and desmethylselegiline from Chinoin Pharmaceutical and Chemical Works (Budapest, Hungary). l-Methamphetamine and l-amphetamine were obtained from Sigma-Aldrich (St. Louis, MO).
Determination of Selegiline and Its Metabolites. Selegiline and its metabolites were analyzed by gas chromatography using nitrogen-selective detection. The assay method is described in detail elsewhere (Taavitsainen et al., 2000). Briefly, samples were made alkaline by adding potassium hydroxide and extracted with heptane containing 2% (v/v) isoamyl alcohol. A portion of the extract was injected into the gas chromatograph and run using a temperature gradient and helium as the carrier gas. Peaks were detected with a nitrogen-phosphorus detector. Peak areas were used for quantitation.
Modeling. Molecular modeling of mammalian microsomal P450s using the CYP102 bacterial crystal structure as a template has been carried out for all of the major P450 families associated with the phase 1 metabolism of drugs and other foreign compounds (Lewis et al., 1999). Three-dimensional models of human P450s have been constructed from the CYP102 hemoprotein domain, for which the X-ray coordinates are known, both in the substrate-bound (Li and Poulos, 1997) and substrate-free states (Ravichandran et al., 1993). The methodologies employed in homology modeling of P450 enzymes from CYP102 are described in detail elsewhere, including a discussion of the rationale for using the CYP102 structure as the preferred modeling template (Lewis, 1996). A number of potential probe substrates (25 in total) for various human P450 enzymes have been identified and tested in the models for CYPlA1, CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 (Lewis et al., 1999). Selegiline was tested in these models.
In Vitro Assays.cDNA-expressed enzymes. Yeast expressing one of the following enzymes, CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4, were produced in the Laboratory of Molecular Toxicology, Institute of Environmental Medicine, Karolinska Institute, Stockholm, Sweden. The expression levels were relatively high, 30 to 200 pmol/mg of microsomal protein. All microsomes produced good difference spectra showing a homogenous peak at 450 nm in the reduced carbon monoxide-bound state of the hemoprotein with no signs of P420. The catalytic properties of the yeast microsomes were evaluated using probe substrates, and all were active with affinities for the substrates as expected (Andersson et al., 2001).
Selegiline (1 mM) was incubated with 0.2 mg of yeast microsomal protein in 0.1 M potassium phosphate buffer, pH 7.4, containing 1 mM NADPH in a total volume of 0.2 ml at 37°C. The reactions were started by the addition of NADPH after a 2-min preincubation and terminated after 20 min by adding 20 μl of 2 M NaOH. Incubations with CYP3A4 were also carried out in the presence of additional human cytochrome P450 reductase and cytochrome b5 (1:1:3 ratio), which were added to the yeast microsomes and placed on ice 10 min before starting the incubation.
In subsequent experiments, conditions were used which ensured linearity with protein and time. Substrate concentrations of between 0–640 μM were used for determination of the Michaelis-Menten kinetics.
Human Liver Microsomes.Origin of livers and preparation of microsomes. Human liver samples used in this study were obtained from kidney transplantation donors. The age of the donors (comprising both males and females) ranged from 7 to 71 years. All samples were histologically normal. None of the subjects was receiving any drugs known to affect the activity of enzymes of drug metabolism. Information on cigarette smoking and alcohol ingestion was not available for all subjects. The use of tissue surplus to requirement was approved by the appropriate ethics committee in each university. Microsomes were prepared according to standard procedures. The final microsomal pellet was suspended in 0.1 M phosphate buffer to a concentration of approximately 20 mg of protein/ml. Protein concentration was measured by the method of Bradford (1976). The livers were thoroughly characterized for their P450 apoprotein content and specific model activities, and were shown to contain all expected P450 enzymes, which were inhibitable by P450-specific chemical inhibitors. Information on apoprotein content of the samples can be found in Edwards et al. (1998). The samples used in the present study correspond to sample numbers 2, 5, 6, 11, 12, 14, 20, 23, and 24. All activities were within the normal ranges reported previously (Boobis et al., 1998).
Inhibition by Selegiline of P450-Specific Enzyme Activities. The following enzyme assays were employed: ethoxyresorufin O-deethylation (CYP1A1/2) (Burke et al., 1977), coumarin 7-hydroxylation (CYP2A6) (Aitio 1978), with a slight modification as described by Raunio et al. (1988, 1990), bupropion hydroxylation (CYP2B6) (Faucette et al., 2000; Hesse et al., 2000), tolbutamide methylhydroxylation (CYP2C9) (modified from Knodell et al., 1987 and Sullivan-Klose et al., 1996), S-mephenytoin 4′-hydroxylation (CYP2C19) (Wrighton et al., 1993), dextromethorphan O-demethylation (CYP2D6) (modified from Park et al., 1984 and Kronbach et al., 1987), chlorzoxazone 6-hydroxylation (CYP2E1) (Peter et al., 1990), and testosterone 6β-hydroxylation (CYP3A4/5) (Waxman et al., 1983). The incubation conditions were similar for the different reactions. If not otherwise stated, each analytical method was applied according to the reference mentioned.
IC50 values were determined by adding selegiline at final concentrations of 0.1, 1,10, 100, and 1,000 μM to the incubation mixture. The resultant activities were compared with those from control incubations into which only solvent (water) had been added. The IC50 values (the concentration of inhibitor causing 50% inhibition of the original enzyme activity) were determined graphically by linear regression analysis of the plot of the logarithm of inhibitor concentration versus percentage of activity remaining after inhibition using Origin, version 4.10 (OriginLab Corp., Northampton, MA). The reference inhibitors were furafylline for CYP1A2, methoxsalen for CYP2A6, sulfaphenazole for CYP2C9, omeprazole for CYP2C19, quinidine for CYP2D6, pyridine for CYP2E1, and ketoconazole for CYP3A4. Methoxsalen and omeprazole are known also to inhibit P450s other than their targets, but there are no selective reference inhibitors for CYP2A6 and CYP2C19 (Pelkonen et al., 1998).
In Vitro Incubation System for Selegiline Metabolism. Selegiline (20–200 μM) was incubated for up to 2 h at 37°C with human liver microsomes in an incubation system that comprised human liver microsomes (pooled sample from 10 livers; 0.25 mg), NADPH (1.2 mM) or NADPH-regenerating system (glucose-6-phosphate dehydrogenase; 4 mM NADP), and phosphate buffer, pH 7.4, in a final volume of 1 ml. Formation of the metabolites, DMS and MA, was linear for up to 0.75 h.
Inhibition of microsomal metabolism of selegiline by P450-specific inhibitors. Inhibition of selegiline metabolism in human liver microsomes by various P450-specific inhibitors was studied in the incubation system described above (20 μM selegiline; incubation time, 30 min). The following inhibitors were used: CYP1A2, furafylline (1, 2, or 5 μM); CYP2A6, coumarin (10, 20, or 50 μM); CYP2C8, quercetin (3, 10, 30 μM); CYP2C9, sulfaphenazole (3, 10, or 30 μM); CYP2C19, S-mephenytoin (100, 200, or 500 μM); CYP2D6, quinidine (1, 5, or 20 μM); CYP2E1, pyridine (1, 5, or 20 μM); and CYP3A4, ketoconazole (1, 5, or 20 μM). All inhibitors were added in methanol (1%) except pyridine, which was added in water.
Correlation study. Selegiline was incubated using the system described above, and metabolism was determined in nine different human liver samples, which had previously been characterized for their content and activity of all of the major drug-metabolizing forms of P450 (CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, CYP3A5, and CYP4A11). The formation rates of desmethylselegiline and methamphetamine were correlated with P450 specific activities, and the amounts of P450 apoproteins were quantified by Western immunoblotting using specific antipeptide antibodies, as described previously (Edwards et al., 1998).
Human Hepatocytes.Isolation and culture of human hepatocytes. Two approaches were tested. In the first, hepatocytes used for the present work were obtained from the resection of an oxalic liver of a young female and were seeded in Williams medium supplemented with 10% fetal calf serum at a density of one million cells in 2 ml of medium in a Petri dish, 2.5 cm in diameter. They showed a viability of >65%. Cells were maintained in a medium containing no serum but supplemented with 5 × 10-5 M hydrocortisone. Medium was refurbished daily (Guguen-Guillouzo and Guillouzo, 1986; Abdel Razzak et al., 1993). Since the cells seemed to have low metabolic capacity, they were only used for qualitative purposes.
In the second approach, surgical liver biopsies (weighing 1–3 g) were obtained from patients undergoing cholecystectomy after informed consent was obtained. Patients had no known liver pathology, nor did they receive medication during the weeks prior to surgery. None of the patients was a habitual consumer of alcohol or other drugs. In total, four liver biopsies were used. Patients' ages ranged from 62 to 82 years. Hepatocytes were isolated using a two-step tissue microperfusion technique as described elsewhere (Gómez-Lechón et al., 1997). Cellular viability, estimated by dye exclusion test with 0.4% trypan blue in saline, was greater than 90%. Hepatocytes were seeded on fibronectin-coated plastic dishes (3.5 μg/cm2) at a density of 8 × 104 viable cells/cm2 and cultured in Ham's F-12/Lebovitz L-15 (1:1) medium supplemented with 2% newborn calf serum, 10 mM glucose, 50 mU/ml penicillin, 50 μg/ml streptomycin, 0.2% bovine serum albumin, and 10 nM insulin. One hour later, the medium was changed, and after 24 h, cells were shifted to serum-free, hormone-supplemented medium (10 nM insulin and 10 nM dexamethasone). The medium was changed daily. Under these culture conditions, cells are metabolically competent (Donato et al., 1995; Gómez-Lechón et al., 1997).
Cytotoxicity Assays. Cytotoxicity was assessed to assure the cells' viability and metabolic competence under the conditions used in further incubations. Hepatocytes were seeded on 96-well microtiter plates, and treatment with increasing concentrations of selegiline started at 24 h of culture. Cells were exposed to the compound for the following 48 to 72 h, and the compound was added daily at the time of medium renewal. Cellular viability was assessed at the end of the treatments by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) test as described in detail elsewhere (Borenfreund et al., 1988). The IC50 and IC10 values, representing the concentrations of the compound that reduce viability by 50% and 10% with respect to the controls, were determined from the dose-response curves obtained. The IC50 value was ≤0.125 mM.
7-Ethoxycoumarin O-Deethylase Activity Assay. 7-Ethoxycoumarin O-deethylation is catalyzed by several P450 enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8-9, CYP2E1, and CYP3A4/5; Waxman et al., 1991). Enzyme activity was measured in intact cells (Gómez-Lechón et al., 1997). Cell monolayers were washed twice with warm PBS, and the assay was initiated by adding 800 μM 7-ethoxycoumarin to the culture medium. Cells were incubated for 30 min at 37°C, and the reaction was stopped by aspirating the incubation medium from the plates. To hydrolyze any 7-hydroxycoumarin conjugates, 1 ml of the medium was incubated with β-glucuronidase/arylsulfatase (200 Fishman units/1200 Roy units) for 2 h at 37°C. Hydrolysis was stopped by adding 125 μl of 15% trichloroacetic acid and 2 ml of chloroform, and then shaking the mixture for 10 min at 37°C. After centrifugation (2,000g for 10 min), the organic phase was extracted with 1 M NaOH, and the fluorescence of the aqueous phase was measured (excitation 368 nm, emission 456 nm) in a microplate fluorimeter. Activity was expressed as picomoles of 7-hydroxycoumarin formed per minute and per milligram of cellular protein assessed by the Lowry method (Lowry et al., 1951).
Incubation of Human Hepatocytes with Selegiline. For metabolic studies, hepatocytes from biopsies were incubated with 62.5 and 125 μM selegiline and hepatocytes from surgical resections with 100 and 500 μM selegiline. Treatment was started at 24 h of culture. Medium and cells were subsequently frozen after 10, 24, and 48 h of continuous incubation with the compound, to be analyzed for selegiline and its metabolites. Samples from parallel plates without cells, incubated with medium containing the compound, were also collected after the same incubation periods.
Preparation and Culture of Precision-Cut Human Liver Slices. The sources of the tissue culture materials were as described previously (Beamand et al., 1993; Lake et al., 1998). Samples of human liver (surplus to transplant requirements) were collected and transported to BIBRA on ice. Tissue cylinders from liver samples were prepared using a 10-mm-diameter motor-driven tissue coring tool. From the cylinders, liver slices (200–300 μm) were prepared in oxygenated (95% O2/5% CO2) Earle's balanced salt solution containing 25 mM d-glucose, 50 μg/ml gentamicin, and 2.5 μg/ml fungizone using a Krumdieck tissue slicer (Alabama Research and Development Corporation, Munford, AL). The liver slices were floated onto Vitron Inc. (Tucson, AZ) type C titanium roller inserts (two slices per insert) and cultured in glass vials containing 1.7 ml of culture medium in a Vitron dynamic organ incubator. The culture medium consisted of RPMI 1640 medium containing 5% (v/v) fetal calf serum, 0.5 mM l-methionine, 1 μM insulin, 0.1 mM hydrocortisone 21-hemisuccinate, 50 μg/ml gentamicin, and 2.5 μg/ml fungizone. Liver slice cultures were maintained at 37°C in an atmosphere of 95% O2/5% CO2. After 30 min, the medium was changed to fresh serum-free RPMI 1640 medium containing 0.5 mM l-methionine, 1 μM insulin, 0.1 mM hydrocortisone 21-hemisuccinate and 0 to 500 μM selegiline (dissolved directly in the culture medium). Liver slices were incubated for periods of 30 to 90 min, and the incubations were terminated by removing the vials from the incubator and plunging them into ice. Appropriate blank incubations (i.e., liver slices in medium without any selegiline and selegiline in medium without any liver slices) were also performed. To achieve a good recovery of parent compound and metabolites (Worboys et al., 1995), the liver slices were removed from the mesh of the roller inserts and homogenized in the culture medium by sonication (Beamand et al., 1993). Liver slice/medium homogenates were assayed for total protein content by the method of Lowry et al. (1951) employing bovine serum albumin as standard. Total protein content was determined to allow for any differences in liver slice thickness between vials and to provide a scaling factor for intrinsic clearance calculations. The liver slice/medium homogenates were stored at -80°C prior to dispatch to the laboratory undertaking the analysis of selegiline metabolites.
Calculations.Scaling from in vitro candidate systems to whole liver. Intrinsic clearance (CLint) is defined as the proportionality constant between the initial rate of the enzymatic reaction (V0) and the drug concentration (C). According to the Michaelis-Menten equation, V0/C = Vmax/Km + C = CLint. At low substrate concentrations, the equation reduces to CLint = Vmax/Km (Ito et al., 1998).
In all in vitro systems, either the rate of disappearance of the parent compound or the rate of the appearance of metabolites was used to determine Km and Vmax (or V0) values. It was not always strictly possible to use initial linear conditions, but the deviations were modest. No allowance was made for any nonspecific loss of selegiline or its metabolites in the incubations. In all systems, appropriate scaling factors were required to convert the primary kinetic data (Vmax/Km or intrinsic clearance) to whole human liver (hepatic intrinsic clearance). The scaling was somewhat different for each in vitro system.
For each cDNA-expressed P450, the form-specific intrinsic clearance was calculated separately. Then, the calculated value was corrected by the human liver microsomal content of that particular form based on results of immunochemical quantification, as published by BD Gentest at http://www.gentest@bd.com. These values are the average content of each form found in 12 different human livers and are as follows: CYP1A2, 50 pmol/mg; CYP2A6, 66 pmol/mg; CYP2B6, 26 pmol/mg; CYP2C9, 55 pmol/mg; CYP2C19, 26 pmol/mg; CYP2D6, 11pmol/mg; CYP2E1, 53 pmol/mg; and CYP3A4, 127 pmol/mg, and upscaled assuming 45 mg of microsomal protein/g of liver (Houston and Carlile, 1997; Carlile et al., 1997) and a liver mass of 1,500 g.
In liver microsomes, the intrinsic clearance was converted to hepatic clearance by assuming 1 g of liver contains 45 mg of microsomal protein and the liver mass to be 1,500 g.
In liver slices, scaling was performed on the basis of the liver slice whole homogenate protein content compared with human liver total protein content. Human liver whole homogenate protein has been determined by homogenizing samples of the liver used to prepare the liver slices and determining the protein content by the method of Lowry et al. (1951). Human liver total protein content of 200 mg of protein/g of liver wet weight has been used in the literature (Bayliss et al., 1990).
In hepatocytes, whole liver clearance was calculated assuming that cell yield was 120 × 106 cells/g of liver (Iwatsubo et al., 1997).
Scaling from liver to in vivo. To calculate the hepatic organ clearance, the well stirred model (Wilkinson and Shand, 1975) excluding protein binding, suggested by Obach et al. (1997) to give a more accurate estimate, was applied. A liver blood flow, QH = 1.450 l/min (Davies and Morris, 1993), was used. According to the model, the organ clearance is CLH = QH · CLint/QH + CLint.
Results
Homology Modeling. Selegiline is a tertiary amine bearing an N-methyl group. This indicates N-demethylation as a likely metabolic pathway for the compound by P450 enzymes. Rotation of the C-N bond could also enable N-depropargylation. Accordingly, homology modeling indicated that the major biotransformations of selegiline would occur by dealkylations of the side chain.
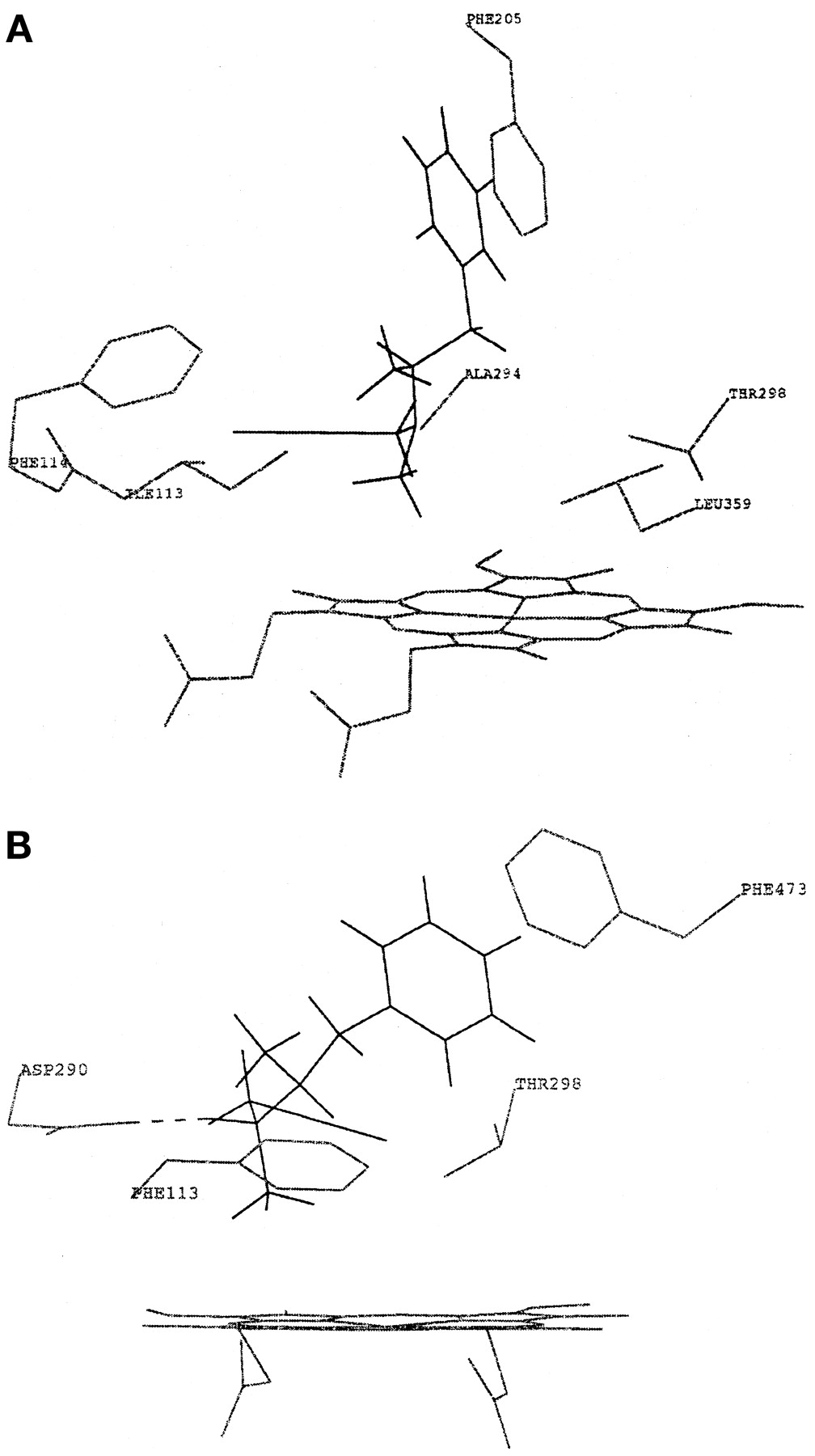
Modeling of selegiline in CYP2B6 indicated its similarity with the likely mode of binding for benzphetamine, which is also N-demethylated by the enzyme. As shown in Fig. 2A, the substrate is able to become oriented within the putative active site of the CYP2B6 such that the N-methyl group lies close to the heme iron at a distance of 3.6 Å. Orientation of selegiline for N-demethylation is effected by a combination of favorable interactions with complementary amino acid residues, essentially hydrophobic in nature, which are within the heme environment. Interactions are with Phe205 (π-stacking) and with Ile113, Phe114, and Ala294 (hydrophobic). All four residues have been the subject of site-directed mutagenesis experiments in CYP2B subfamily proteins, and typical CYP2B6 substrates bind to the same residues.

The modeled P450 interaction of selegiline (black) shows the orientation of the molecule within the putative active site (gray) of CYP2B6 favoring N-demethylation (A) and of CYP2D6 favoring N-depropargylation (B).
CYP2D6 was also predicted to participate in the metabolism of the compound (Fig. 2B). According to the model, the enzyme would catalyze predominantly the N-depropargylation of selegiline to methamphetamine. Interaction by CYP2D6 inhibition could also be possible. Interactions are with Asp290 (ion-pairing), Phe473 (π-stacking), and Phe113 (hydrophobic). Of these, the first two have been mutated in CYP2D6 and have been shown to affect substrate binding. Also, typical substrates of the enzyme bind to the same residues. Modeling for CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, and CYP3A5 suggested no other form participating in the metabolism of selegiline.
Recombinant Expressed Enzymes. All nine available recombinant P450s were evaluated as catalysts of the metabolism of selegiline, using a 200 μM substrate concentration, including kinetic analysis with respect to time and protein concentration. Selegiline was metabolized by CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, and CYP2E1 (Fig. 3). CYP2D6 showed hardly any activity. CYP3A4 was not active without added human P450 reductase and cytochrome b5, which increased the formation of desmethylselegiline about 5-fold and methamphetamine formation 2- to 5-fold, depending on the substrate concentration. Both metabolites were produced by all forms of P450 that were active, and the rate of MA formation generally exceeded that of DMS. No amphetamine was formed by any of the forms tested.
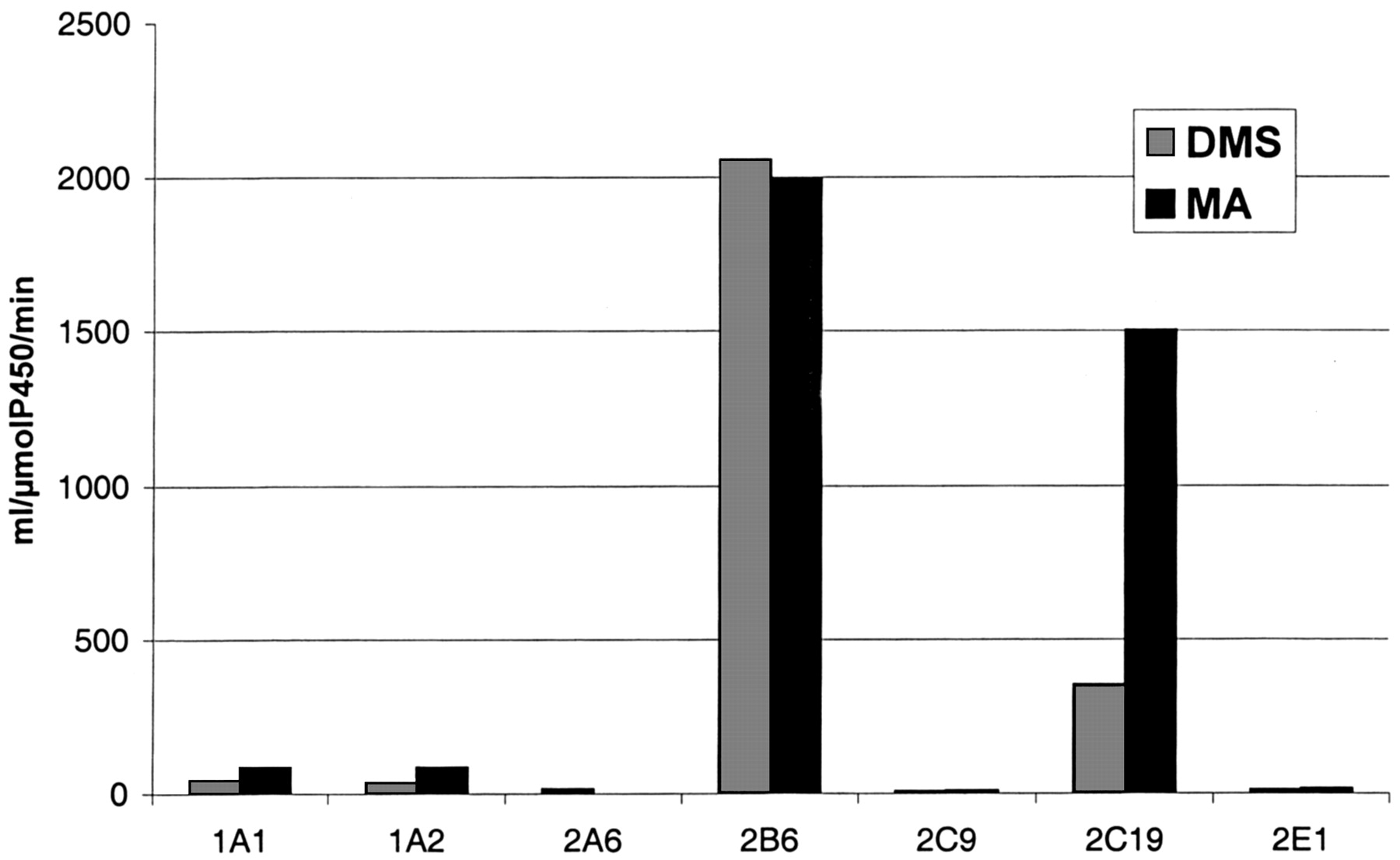
Intrinsic clearances for individual recombinant P450s calculated on the basis of DMS or MA formation from selegiline.
No measurable activity was found for CYP2D6 for either DMS or MA formation.
CYP2B6 and CYP2C19 were the main forms responsible for the formation of metabolites, as shown by their intrinsic clearances, i.e., Vmax/Km ratios, for both desmethylselegiline and methamphetamine production (Fig. 3). In the case of CYP2B6, the ratio was almost the same for the formation of both metabolites, whereas in the case of CYP2C19, the ratio for MA production was 3 times higher than that for DMS formation. When the relative amounts of the P450 enzymes in human liver (see Materials and Methods) were taken into account, the contributions of CYP2B6 and CYP2C19 to the overall clearance of selegiline were calculated to be 52.1% and 34.5% of the total, respectively. In contrast, the role of other P450 enzymes was estimated to be minor. When the contribution of all six active forms, excluding CYP3A4, to the total hepatic clearance were summed, a value of 223 l/h was obtained. Studies on selegiline metabolism with cDNA-expressed P450 enzymes, to reveal the contribution by CYP3A4, have been continued after the completion of the present work (Hidestrand et al., 2001).
Human Liver Microsomes.Metabolite pattern and metabolic rates. The biotransformation of selegiline in microsomes was assessed both from substrate disappearance and from product formation. Metabolites could be detected at all substrate concentrations tested (20, 50, and 200 μM). Substrate turnover was appreciable at all concentrations, up to 200 μM, with no evidence of saturation (Fig. 4). Two metabolites, DMS and MA, were readily detected, with trace amounts of a third (amphetamine) evident at the highest substrate concentration (200 μM) after 60 min. Levels of this metabolite were at the limits of detection of the assay. Formation rates of the two major metabolites increased with time for up to 45 min, after which they leveled off. Formation rates of DMS and MA showed some evidence of beginning to saturate at 200 μM selegiline (Fig. 4). These data were used to obtain approximate estimates of the kinetic constants for the formation rates of the two metabolites by nonlinear regression analysis.

Hepatic microsomal metabolism of selegiline.
Substrate disappearance (▪), MA formation (▾), and DMS formation (▴) are shown. Results are from a pool of liver samples (n = 12) and are means of replicate determinations.
The apparent Km values for both DMS (99 μM) and MA (103 μM) formation in microsomes were the same. In contrast, the maximum velocity for MA formation (1551 pmol/min per mg of microsomal protein) was about 2-fold greater than that for DMS formation (733 pmol/min per mg of microsomal protein). Consequently, the calculated intrinsic clearances were 15.1 μl/min per mg of microsomal protein for MA formation and 7.4 μl/min per mg of microsomal protein for DMS formation, respectively. These values scale up to a total hepatic clearance of 61.2 l/h for MA and 30.0 l/h for DMS, and a total metabolic CL via these two pathways of 91.2 l/h (Table 3).
Estimates of the metabolic rate in various in vitro systems
Inhibition of P450-Specific Model Activities in Human Liver Microsomes. Selegiline was an inhibitor of CYP2B6 (IC50 = 26 μM) and CYP2C19 (IC50 = 28–37 μM) and a moderate inhibitor (IC50 = 50–75 μM) of three other forms, CYP2D6, CYP2A6, and CYP1A2, in decreasing order of potency. It had little or no effect on CYP2C9 or CYP3A4 (Table 1). Although differences between the values measured by two laboratories may be due to specific details and conditions between model assays and tissues available, the overall picture of inhibitory potencies was rather similar. The discrepancy between IC50 values of the two CYP2D6-catalyzed model activities, dextromethorphan O-demethylase and debrisoquine hydroxylase, is probably due to kinetic properties of two different substrates.
Effect of selegiline on P450-model activities in human liver microsomes Model substrates and reference inhibitors are listed in parentheses.
Inhibition of Microsomal Metabolism of Selegiline by P450-Specific Inhibitors. Model inhibitors differentially affected the formation of the main metabolites of selegiline (Table 2). Both furafylline and fluvoxamine inhibited an appreciable component of DMS formation. Also, methoxsalen inhibited the formation of DMS, but at much higher concentrations than needed for the inhibition of CYP2A6. Fluvoxamine inhibited the formation of MA. Ketoconazole (CYP3A4) and quercetin (CYP2C8) were the most effective inhibitors of selegiline metabolism in human liver microsomes, causing 58% and 47% inhibition of DMS formation and 71% and 55% inhibition of MA formation, respectively. Quinidine, a CYP2D6 selective inhibitor, had a very minor inhibitory effect on selegiline metabolism.
Inhibition of microsomal metabolism of selegiline by P450-specific inhibitors
Correlation Study. Several significant correlations were found for selegiline metabolism in nine different human liver samples characterized for their major P450 content. Formation of the two major metabolites was correlated with total P450 content (DMS: r2 = 0.58, P < 0.02; MA: r2 = 0.67, P < 0.01). DMS formation was significantly correlated with MA formation (r2 = 0.83, P < 0.001), suggesting the participation primarily of the same P450 enzymes in their formation. Both DMS (r2 = 0.64, P < 0.01) and MA (r2 = 0.77, P < 0.002) production correlated with phenacetin O-deethylase activity (CYP1A2). Less marked, but still significant, correlations were found between MA formation and CYP2C19 (r2 = 0.52, P < 0.05) and between DMS formation and CYP2C9 (r2 = 0.52, P < 0.05). DMS formation showed a significant correlation with CYP2B6 content (r2 = 0.56, P < 0.05), and formation of both metabolites showed a significant correlation with CYP2C8 content (DMS: r2 = 0.69, P < 0.01; MA: r2 = 0.62, P < 0.02). MA formation was correlated with microsomal CYP1A2 content (r2 = 0.49, P < 0.05).
Primary Cultured Human Hepatocytes.Cytotoxicity and induction potential of selegiline. Selegiline was incubated with human hepatocytes at concentrations from 30 to 1,000 μM for 48 to 72 h, to determine its cytotoxicity by the MTT assay. Under these conditions, the compound showed minimal cell toxicity at concentrations below 125 μM.
The potential P450 inducing effects of selegiline were evaluated after 48–72 h incubation, by measuring 7-ethoxycoumarin O-deethylase activity. Selegiline did not increase enzyme activity. Rather, the drug caused a dose-dependent inhibition (40–60% reduction) of 7-ethoxycoumarin O-deethylase activity.
Rate of Elimination and Metabolite Production. Two concentrations of selegiline (62.5 and 125 μM) were incubated for 10, 24, or 48 h with human hepatocytes originating from surgical biopsies. The cells produced considerable amounts of DMS and MA. In several experiments, the formation rate of MA was 2- to 3-fold greater than the formation rate of DMS. Amphetamine was formed to a lesser extent. Total recovery of the compounds (SEL + DMS + MA + A) from the incubate clearly decreased with increasing time.
The kinetics of selegiline elimination were evaluated on the basis of substrate disappearance, and an “initial” velocity of 13.7 nmol/h per one million cells was obtained. This value corresponds to a total hepatic clearance of about 16.1 l/h. However, the rate of selegiline metabolism was determined over a 10-h period, and true initial conditions were probably lost within the first hour or two. Clearance estimation based on the sum of metabolite formation yielded a much lower value (Table 3). This may be related to the recovery problems mentioned above.
Both DMS and MA, but not amphetamine, were detected as biotransformation products of selegiline in hepatocytes isolated from surgical liver resections. However, the overall metabolism in these cells was low, and no useful clearance estimate could be calculated. This may also be related to recovery problems, in this particular case.
Precision-Cut Human Liver Slices. Selegiline underwent biotransformation in human liver slices at all substrate levels tested. Both DMS and MA were produced at appreciable rates, and the amounts formed increased with substrate concentration, but no time dependence was observed after 0.5 h of incubation, suggesting that reaction equilibrium had been reached before that point, with some clearance of the primary products by secondary metabolism. Consistent with this, detectable levels of amphetamine were observed, particularly at the highest substrate concentration.
Kinetics were evaluated on the basis of the sum of the formation rates of the two metabolites. The velocity concentration curves (Eadie-Hofstee plot) indicated biphasic kinetics. Apparent kinetic parameters were calculated for the first (high affinity) phase only. This resulted in an apparent Km of 4 μM and Vmax of 14 pmol/min per mg liver slice whole homogenate protein. The corresponding intrinsic clearance was 3.5 μl/min per mg liver slice whole homogenate protein. This scales up to a total (hepatic) metabolic clearance of 36.5 l/h.
Discussion
Elucidation of the Identity of Metabolites and the Metabolic Route. The primary metabolites of selegiline in humans are desmethylselegiline and methamphetamine (Reynolds et al., 1978; Shin, 1997). Amphetamine is a secondary product formed by dealkylation of either of the two compounds (Fig. 1). These three metabolites have been detected in human urine, blood plasma, and serum, as well as in cerebrospinal fluid (Reynolds et al., 1978; Heinonen et al., 1989; Shin., 1997). Methamphetamine is the most abundant metabolite observed in vivo. In vitro, conversion of selegiline to methamphetamine by rat liver microsomes also occurred at a high rate (Yoshida et al., 1986).
In the current study, all of the in vitro systems tested correctly produced the two primary metabolites from selegiline. Since the identity of the test compound was originally not revealed to the participating laboratories, other than to the molecular modeling group, this group was the only one in a position to make any predictions on the structure of the metabolites. Modeling correctly predicted the sites of metabolic attack and the metabolite structures. It predicted both methamphetamine and N-desmethylselegiline as the primary P450 products. Methamphetamine was the most abundant product with cDNA-expressed enzymes, microsomes, hepatocytes, and liver slices, in agreement with the in vivo data.
The secondary metabolite, amphetamine, was not detected when using recombinant enzymes and was produced in only trace amounts by the other systems. This is not surprising in view of the reaction rates of the primary pathways. Hydroxylated amphetamines, which have also been detected as secondary metabolites of selegiline in vivo (Yoshida et al., 1986; Shin, 1997), were not observed in the studies reported here. Since the analytical method used includes extraction into an organic solvent, these compounds could have escaped detection, as a consequence of their polarity. However, no hydroxylated products were found in an earlier study in which selegiline was incubated with rat liver microsomes (Yoshida et al., 1986). Because these products would require the involvement of three sequential reactions, significant amounts would not be anticipated in vitro.
Elucidation of the Metabolic Rate and Prediction of in Vivo Clearance. In humans, selegiline is eliminated exclusively by biotransformation, resulting in a high systemic clearance. The reported clearance, 236 l/h (Heinonen et al., 1994), exceeds the maximum metabolic capacity of two parallel operating hepatic reactions. The theoretical maximum clearance by formation of desmethylselegline and methamphetamine would be equal to twice the hepatic blood flow; i.e., approximately 2 × 90 l/h. Some extrahepatic metabolism of selegiline by P450s has been observed in rats (Yoshida et al., 1987), and it may be that this also contributes to the elimination of the drug in humans. Since the drug concentration in plasma is very low, after an oral dose of 10 mg, the maximum level is approximately 10 nM (Heinonen et al., 1994); binding to monoamine oxidase type B also makes a minor contribution to the observed overall elimination rate.
All of the in vitro techniques indicated that selegiline is readily metabolized by P450 enzymes. For the reasons outlined above, predicted clearances should be compared with the theoretical value of hepatic intrinsic CL rather than the total body CL. Upscaling of the intrinsic clearance resulted in an underestimate of the in vivo clearance for all in vitro systems except the recombinant enzymes where, instead, a slight overestimate was obtained (Table 3). In the case of microsomes, hepatocytes, and liver slices, biotransformations in addition to those quantified may take place. Thus, measurement of substrate disappearance, rather than the formation of individual metabolites, should give a more valid estimate of the drug clearance. However, any nonspecific loss of substrate would bias this estimate. In fact, an acceptable estimate of in vivo clearance was achieved from the sum of the rates of formation of the two main metabolites in human liver microsomes (Table 3). Hepatocytes performed less satisfactorily, most probably because the incubation time was too long, and hence, it was not possible to obtain an accurate estimate of initial rates when the measurements were made at the end of the incubation period. Over this interval, it is very likely that secondary reactions had consumed some of the primary metabolites produced, reducing the accuracy of the estimation of reaction rate and clearance.
The same mechanisms may have influenced, in part, the results obtained with liver slices, in which metabolite formation gave a low estimate of clearance (Table 3), although the incubation time was shorter and hence there would be less opportunity for secondary metabolism. It is possible that with a rapidly metabolized compound such as selegiline, the surface cell layers of the slice exhaust the substrate before it reaches the inner cell layers. Consequently, the observed total metabolic rate will be lower than with a similar mass of freely accessible enzymes, e.g., in microsomes, resulting in a reduced clearance estimate. Differences between surface and inner layers of cells in substrate access may well also explain the observed biphasic kinetics of selegiline metabolism in liver slices.
Indeed, despite the relative underestimation of total in vivo CL, all of the in vitro systems tested successfully classified the compound into the correct clearance category; i.e., all showed selegiline to be a high clearance drug. In addition, it is anticipated that these systems could correctly rank the clearance of a series of closely related drug candidates, inasmuch as this depends on relative rather than absolute predictions. In practice, this result would be sufficient for screening purposes, and more accurate clearance values would be obtained during further drug development.
Identification of the Participating P450 Isoforms. Prediction of pharmacokinetic drug interaction potential as well as of individual variability in drug response relies on the identification of the P450 enzymes involved in the metabolism of the drug. In the case of selegiline, characterization of the metabolizing P450s remains to be completed (see Taavitsainen et al., 2000; Hidestrand et al., 2001). Yoshida et al. (1986), in addition to proving that the conversion of selegiline to methamphetamine in rat liver microsomes is catalyzed by the P450 system, showed that the reaction was inducible by phenobarbital. Thus, enzymes of the CYP2B subfamily (Waxman and Azaroff, 1992) might be involved.
In humans, it has been suggested that CYP2D6 is the enzyme responsible for selegiline demethylation. This was based on findings with human recombinant CYP2D6 microsomes but was not supported by the model of CYP2D6 used by the authors (Grace et al., 1994). They proposed that the discrepancy was explicable by an atypical reaction mechanism. Similar activity of CYP2D6 was reported in another study with microsomes from CYP2D6 cDNA-expressing cells (Bach et al., 2000). In the present study, homology modeling indicated an interaction of selegiline with CYP2D6 that could also lead to inhibition of the enzyme. In the recombinant expressed enzyme system, activity of this form of P450 was below the limit of detection. No support for CYP2D6 participation was obtained in either the microsomal inhibition or correlation studies: quinidine did not significantly inhibit DMS or MA formation (Table 2), and the IC50 of SEL for CYP2D6 was at least 120-fold higher than that of the reference inhibitor (Table 1).
There was no correlation between either CYP2D6 content or activity with the rates of formation of either metabolite. A similar lack of correlation with CYP2D6 was found by Jurima-Romet et al. (2000). In humans in vivo, the CYP2D6 polymorphism was shown to have no effect on the metabolism of selegiline, the kinetics of the compound being very similar in poor and extensive metabolizers of debrisoquine (Scheinin et al., 1998). Hence, overall, it appears very unlikely that CYP2D6 plays any significant role in the biotransformation of selegiline in humans.
Studies with both microsomes and recombinant expressed human P450s suggested a role for CYP2C19 in selegiline metabolism. Clear evidence for a contribution from this P450 form was obtained using recombinant expressed enzymes, CYP2C19 being particularly active in depropargylation to produce MA. Additional evidence for a contribution from CYP2C19 was obtained in studies of the inhibition of model reactions (Table 1), selegiline having a lower IC50 value for this enzyme than the reference inhibitor, and the existence of significant correlation between MA formation and CYP2C19 content. The latter result is consistent with data from recombinant enzymes that CYP2C19 is more active in depropargylation than demethylation of selegiline. Data from studies in vivo in extensive metabolizers and poor metabolizers for CYP2C19 (Laine et al., 2001) support a modest contribution of this enzyme to the metabolism of selegiline, via the depropargylation pathway. Hence, although molecular modeling did not identify CYP2C19 as one of the forms of P450 involved in selegiline metabolism, there is good evidence that this enzyme does contribute to the (primary) metabolism of selegiline in vivo, albeit normally only to a minor extent.
Molecular modeling identified CYP2B6 as one of the forms of P450 involved in selegiline metabolism, particularly via demethylation to produce DMS. Correlation studies with hepatic microsomal fractions also suggested a contribution of CYP2B6 to DMS formation. Selegiline inhibited moderately the hydroxylation of a CYP2B6 substrate, bupropion (Table 1). Recombinant expressed CYP2B6 exhibited both high affinity and a high capacity to metabolize selegiline, with similar kinetic constant for the formation of DMS and MA, thus supporting the participation of CYP2B6 in the formation of these two metabolites. The kinetics of CYP2B6 are such that it is predicted that this would be the major form of P450 involved in its metabolism (present study and Hidestrand et al., 2001). However, CYP2B6 expression and activity are highly variable, with appreciable expression in only 20% of the population (Edwards et al., 1998; Ekins et al., 1998; Ekins and Wrighton, 1999; Gervot et al., 1999). Recent studies have established that much of this variability has a genetic origin (Lang et al., 2001). Hence, the role of CYP2B6 in vivo as a determinant of the kinetics and dynamics of selegiline within the population as a whole remains to be determined.
Studies with human liver microsomes implicated CYP2C8 in the metabolism of selegiline. There were significant correlations between CYP2C8 content and the formation rates of both MA and DMS. Similarly, quercetin, a selective inhibitor of CYP2C8 and CYP1A2 (Dierks et al., 2001), inhibited both microsomal depropargylation and demethylation of selegiline to a greater extent than furafylline, a very selective inhibitor of CYP1A2 (Sesardic et al., 1990; Dierks et al., 2001) Recombinant expressed CYP2C8 exhibited only low activity toward selegiline (Hidestrand et al., 2001). Overall, these data suggest that CYP2C8 may play a minor role in selegiline oxidation.
There was also a significant correlation between CYP1A2 content of hepatic microsomal samples and MA formation. In addition, furafylline inhibited the microsomal formation of both MA and DMS, with a greater effect on MA formation, and recombinant expressed CYP1A2 was active in the formation of both metabolites but, again, was more active in MA formation. Hence, the evidence would suggest a modest contribution of CYP1A2 to the metabolism of selegiline, particularly in MA formation.
There was no indication from molecular modeling for the involvement of CYP3A4 in the metabolism of selegiline. Similarly, there was no correlation between microsomal CYP3A4 content and selegiline metabolite formation, nor did selegiline significantly inhibit CYP3A4-dependent reactions. Recombinant CYP3A4 itself was inactive but, in the presence of human cytochrome P450 reductase and cytochrome b5, showed modest activity in both MA and DMS formation (Hidestrand et al., 2001). Ketoconazole inhibited an appreciable component of selegiline turnover by human liver microsomes, and in an earlier study, selegiline metabolism by human liver microsomes was reduced by CYP3A inhibitors (Wacher et al., 1996). However, although ketoconazole is a relatively selective inhibitor of CYP3A4, at the concentrations used, it also inhibits CYP1A2, CYP2B6, CYP2C8, and CYP2C19 (Dierks et al., 2001; Zhang et al., 2002). Thus, there is little evidence for a significant contribution from CYP3A4 to the biotransformation of selegiline.
Predicted Interaction Potential. From the present data, it is apparent that the metabolism of selegiline in human liver to desmethylselegiline and metamphetamine is a multiple enzyme process, involving at least CYP1A2, CYP2B6, CYP2C19, and possibly CYP2C8. The participation of several different forms of P450 in the elimination of the drug reduces the likelihood of possible drug interactions affecting selegiline clearance in humans. In contrast, selegiline is an effective inhibitor of CYP2C19 and hence could possibly reduce the clearance of drugs dependent on this enzyme for their elimination. However, systemic levels of selegiline remain in the low nanomolar range (Heinonen et al., 1994), and micromolar concentrations would be required to inhibit CYP2C19. Thus, it is highly unlikely that any interaction would arise by this mechanism either. Consistent with these conclusions, no metabolism-related drug interactions involving selegiline have been reported to date.
Conclusions. When comparing the results of the present study to the existing in vivo data in humans (Heinonen et al., 1994; Laine et al., 1999), it can be concluded that, in spite of the variable numeric values for predicted intrinsic clearance, recombinant enzymes, liver microsomes, hepatocytes, and liver slices were all able to predict the metabolic lability, i.e., the high metabolic clearance, of selegiline. Homology modeling correctly predicted the primary metabolic reactions and the sites of metabolism. Indeed, all of the other systems enabled the correct identification of the major metabolic products. Although uniform prediction of the relative contribution of different P450s to desmethylselegiline and metamphetamine formation, the primary metabolic reactions of selegiline, was not obtained, there was a reasonable degree of agreement among them. All of the systems utilized (homology modeling, recombinant enzymes, and microsomes) suggested the participation of several P450 enzymes and (correctly) predicted low interaction potential in vivo. Although none of the techniques studied here was alone able to predict all aspects of the metabolic and kinetic behavior of selegiline in vivo, with an integrated package, all significant characteristics were predictable. It is reasonable to suggest that such an approach would be the best procedure for new chemical entity metabolism screening in general. When a particular aspect of metabolism needs to be investigated further, a single in vitro system, appropriate to the issue of concern, can be selected on the basis of the type of information reported here.
Acknowledgments
We wish to acknowledge the contributions of the other members of the EUROCYP project: Dieter Paul, Rainer Klocke (Fraunhofer Institute, Hannover, Germany), Tommy B. Andersson (AstraZeneca, Mölndal, Sweden), and Gisela Backfisch (Roche Diagnostics, Mannheim, Germany). We also thank Orion Pharma for providing selegiline and its metabolites.
Footnotes
-
↵1 Abbreviations used are: SEL, selegiline; P450, cytochrome P450; MA, methamphetamine; A, amphetamine; DMS, desmethylselegiline; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; CL, clearance.
-
This study was supported by the European Union framework 4 programme (EUROCYP Project BMH4-CT96-0254).
- Received December 3, 2002.
- Accepted May 30, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}