


Abstract
Duloxetine is a potent and balanced dual inhibitor of serotonin and norepinephrine reuptake being investigated for the treatment of depression and urinary incontinence. The disposition of duloxetine was studied in four healthy human subjects after a single 20.2-mg (100.6 μCi) oral dose of [14C]duloxetine in an enteric-coated tablet. The mean total recovery of radioactivity (± S.E.M.) after 312 h was 90.5% (±0.4%) with 72.0% (±1.1%) excreted in the urine. Duloxetine was extensively metabolized to numerous metabolites primarily excreted into the urine in the conjugated form. The major biotransformation pathways for duloxetine involved oxidation of the naphthyl ring at either the 4-, 5-, or 6-positions followed by further oxidation, methylation, and/or conjugation. The major metabolites found in plasma were glucuronide conjugates of the following: 4-hydroxy duloxetine (M6), 6-hydroxy-5-methoxy duloxetine (M10), 4, 6-dihydroxy duloxetine (M9), and a sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7). The major metabolites found in plasma were also found in the urine, but the urine contained many additional metabolites. In addition to duloxetine, 4-hydroxy duloxetine (M14) and an unidentified polar metabolite were observed in feces. Following [14C]duloxetine administration, Cmax was reached at a median of 6 h for both duloxetine and total radioactivity. Duloxetine accounted for less than 3% of the circulating radioactivity based on mean area under the curve values. The elimination half-life of total radioactivity (120 h) was substantially longer than that of duloxetine (10.3 h).
Duloxetine [LY248686, (+)-N-methyl-3-(1-naphthalenyloxy)-2-thiophenepropanamine] is a potent and balanced inhibitor of the reuptake of serotonin (5-hyroxytryptamine, 5HT2) and norepinephrine (NE) in vitro and in vivo (Fig. 1). Duloxetine has demonstrated a relatively evenly balanced and potent inhibition of both the 5HT and NE reuptake at the transport sites and a weak effect on dopamine reuptake in both in vitro and in vivo studies (Pitsikas, 2000; Wong, 1998). Duloxetine lacks significant affinity for muscarinic, histamine H1, α1-adrenergic, dopamine D2, 5HT1A, 5HT1B, 5HT1D, 5HT2A, 5HT2C, and opioid receptors (Wong et al., 1993). The combined action on more than one monoamine neurotransmitter could result in a more favorable clinical outcome when compared with current selective serotonin reuptake inhibitors (Wong and Bymaster, 2002).

Structure of duloxetine.
The asterisk denotes the position of the 14C label.
Preclinical studies in animals have shown that duloxetine enhances the release of 5HT and NE in limbic areas of the rat brain (Rueter et al., 1998) and, under irritated bladder conditions in cats, increases bladder capacity and periurethral striated sphincter electromyographic activity (Thor, 1995). Based upon these data, duloxetine is being studied clinically for use in the treatment of major depressive disorders and stress urinary incontinence. Not only has duloxetine shown efficacy in these two disorders, but it has been shown to be safe and well tolerated (Sharma et al., 2000; Detke et al., 2002; Goldstein et al., 2002; Norton et al., 2002). At doses of 20, 30, or 40 mg b.i.d. administered to healthy male subjects, the mean oral clearance, apparent volume of distribution, and half-life for duloxetine were 114 l/h, 1943 liters, and 12.5 h, respectively (Sharma et al., 2000).
The safety and pharmacokinetics of duloxetine have been evaluated extensively in healthy subjects. This study was conducted in four healthy participants to understand the adsorption, disposition, metabolism, and excretion of duloxetine following a single oral dose of duloxetine hydrochloride in an enteric-coated tablet.
Materials and Methods
Reference Compounds and Chemicals. The following nonlabeled and labeled compounds were synthesized at Eli Lilly and Company: duloxetine HCl, [14C]duloxetine HCl, and 13CD3 duloxetine HCl; 4-, 5-, and 6-hydroxy duloxetine (M14, M12, and M13); N-desmethyl duloxetine (M23); 6-hydroxy-5-methoxy duloxetine (M15); 5-hydroxy-6-methoxy duloxetine (M16); thienyl alcohol (M26); dihydrodiol duloxetine (M2); 4,6-dihydroxy duloxetine (6); glucuronide conjugate 4,6-dihydroxy duloxetine (M9); glucuronide conjugate of 6-hydroxy-5-methoxy duloxetine (M10); sulfate and glucuronide conjugates of 5-hydroxy-6-methoxy duloxetine (M7, M3); glucuronide conjugates of 4- and 6-hydroxy duloxetine (M6, M8); and glucuronide conjugate of dihydrodiol duloxetine (M1). Reagents and solvents were of analytical grade and were obtained from commercial sources.
Synthesis of Primary Metabolites of Duloxetine. The syntheses of some of the duloxetine metabolites M14 to M16 were accomplished by condensing the thiophene side chain 1 with the corresponding fluoronaphthols, 2 to 5 (Fig. 2). The hydroxyl groups were protected as ketals or as acetals, under the conditions for the synthesis of duloxetine (R1 = R2 = R3 = H) (Wheeler et al., 1995). The protecting groups were then removed by acetic acid. The glucuronide conjugates M6 and M10 were synthesized by O-alkylation of M14 and M15 with acetobromo-α-d-glucuronic acid methyl ester followed by saponification of the ester group. Compound M9 was synthesized by enzymatic glucuronidation of 6. The glucuronidation was accomplished by addition of 6 to two cultures, Streptomyces sp. and Actinoplanes missouriensis, which were prepared with expression (bioconversion) medium before incubation. The cultures were incubated for 3 days at 30°C with 165-rpm shaking before filtration and isolation. The sulfate conjugate, M7, was synthesized as shown in Fig. 2. [NMR spectra (in ppm): Compound M13 (in CD3OD): 8.2 (d, 1 H), 7.38 (d, 1 H), 7.28 (d, 1 H), 7.03 (dd, 1 H), 7.01 (d, 1 H), 6.52 (s, 2 H), 5.66 (t, 1 H, methine next to thiophine), 2.85 (m, 2 H, CH2N), 2.42 and 2.25 (m, 1 H each, CH2), 2.42 (s, 3 H, NCH3). Compound M6 (in CD3OD/D2O): 8.33 (dd, 1 H), 8.18 (dd, 1 H), 7.50 (dd, 2 H), 7.30 (d, 1 H), 7.10 (dd, 2 H), 6.95 (dd, 1 H), 6.81 (d, 1 H), 5.75 (d, 1 H methine next to thiophene), 4.97 (d, 1 H of acetal on sugar ring), 3.50 to 3.80 (cluster, 4 H on sugar ring), 3.21 (m, 1 H, CH2N) and 3.10 (m, 1 H, CH2N), 2.70 (s, 3 H, NCH3), 2.49 (m, 1 H) and 2.39 (m, 1 H, CH2). Compound M10 (in CD3OD/D2O): 8.05 (1 H, d), 7.59 (d, 1 H), 7.44(d, 1 H), 7.31 (d, H), 7.29 (d, 1H), 7.10 (d, 1 H), 6.86 (dd, 1 H), 6.83 (d, 1 H), 5.82 (t, 1 H, methine next to thiophene), 5.12 (d, 1 H of acetal on sugar ring), 3.96 (s, 3 H, OCH3), 3.60 to 3.80 (cluster, 4 H on sugar ring), 2.75 and 2.70 (m, 1 H each, CH2N), 2.41 and 2.22 (m, 1 H each, CH2), 2.32 (s, 3 H, NCH3). Compound M7 (CD3OD/D2O): 8.0 (d, 1 H), 7.6 (d, 1 H), 7.26 (d, 1 H), 7.15 (m, 2 H), 6.97 (d, 1 H), 6.80 (dd, 1 H), 6.68 (d, 1 H), 5.68 (t, 1 H methine next to thiophene), 3.80 (s, 3 H, OCH3), 2.60 and 2.52 (m, 1 H each, CH2N), 2.25 and 2.05 (m, 1 H each, CH2), 2.15 (s, 3 H, NCH3).
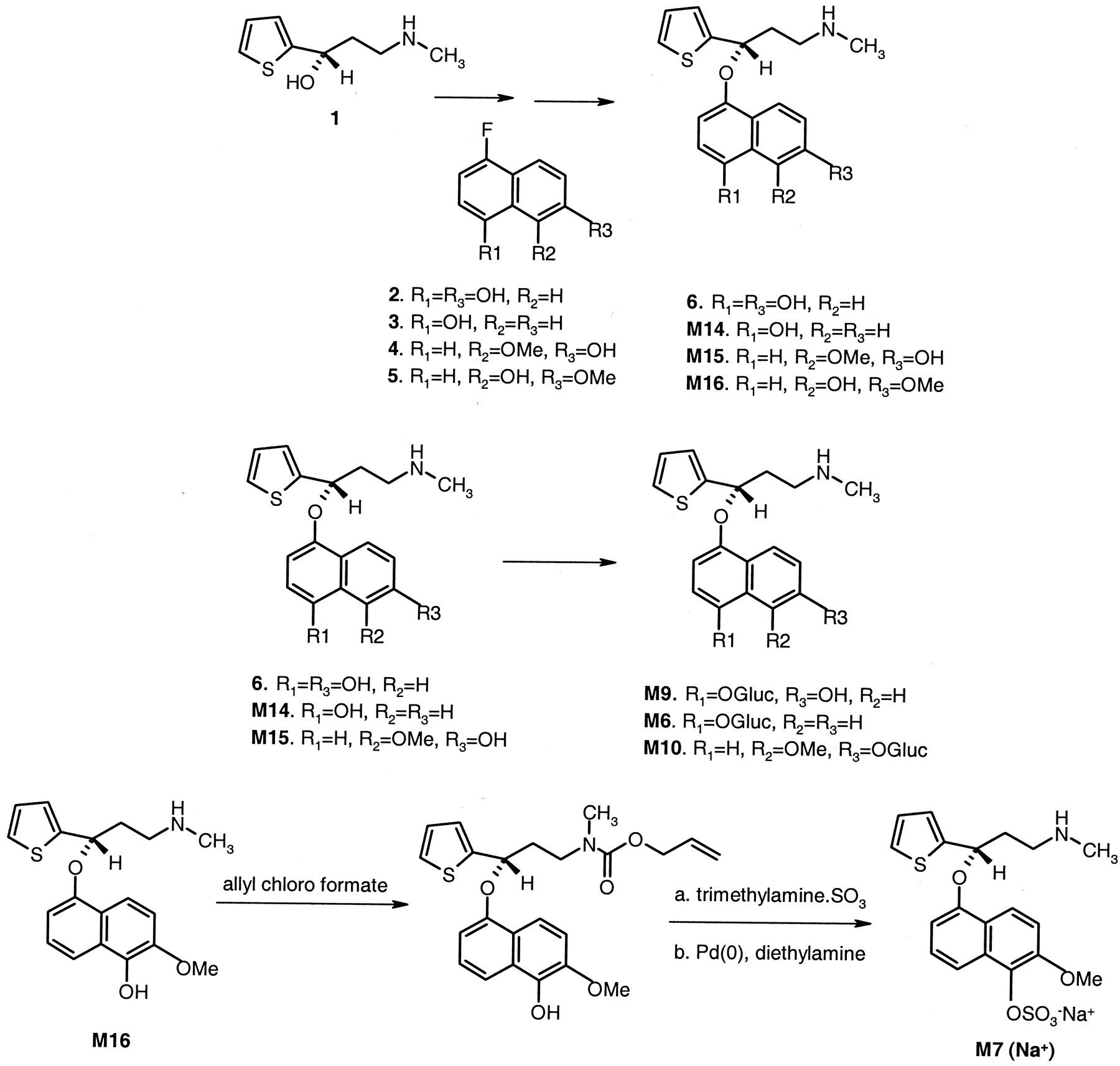
Synthesis scheme for the metabolites of duloxetine.
Study Design. This open-label, single-dose metabolic inpatient study was conducted with four participants (three males and one female) ranging in age from 44 to 48 years (Table 1). Study participants were selected on inclusion/exclusion criteria, medical history, physical examination, and other procedures outlined in the protocol. Before the study started, an institutional review committee approved the protocol and the informed consent document. Study participants gave written informed consent prior to the study. Participants were not involved in the administration of an investigational new drug or a radioactive investigational new drug within 30 days or 12 months, respectively, prior to the start of the study. Prior to dosing, the participants were also tested to determine their metabolic phenotype to CYP2D6 to determine whether they were poor metabolizers or extensive metabolizers. The study was conducted at Lilly Laboratories for Clinical Research, Indiana University Hospital and Outpatient Center, Indianapolis, Indiana.
Subject demographics
Dose Administration. [14C]Duloxetine hydrochloride was synthesized at Eli Lilly and Company (Wheeler et al., 1995), and the 14C label was located at the chiral center of the molecule. Purity of the unlabeled and labeled drug was >99%.
Study drug was supplied as an enteric-coated tablet formulation containing 20.2 mg of unlabeled and radiolabeled duloxetine with an activity of approximately 100.6 μCi. The specific activity of the material was approximately 5 μCi/mg. The dose was administered with 180 ml of water. The subjects fasted from 12 midnight the night before drug administration, and they fasted for an additional 4 h after dosing.
Biologic Sample Collection. Blood samples (20 ml) were drawn from each subject within 0.5 h before dosing (0-h sample), and at approximately 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, 36, 48, 72, 96, 120, 144, 192, 216, and 240 h after dosing in Vacutainer tubes containing sodium heparin. Plasma samples were obtained after centrifugation and aliquots were analyzed for radioactivity. The plasma samples were stored at approximately -20°C for future analyses of duloxetine and metabolites. Since it had been shown previously that radioactivity does not partition into the cellular fraction of human blood, whole blood was not analyzed for radioactivity in this study.
Urine samples were collected in plastic containers. They were collected prior to dosing and at the following time points: 0 to 4, 4 to 8, 8 to 16, 16 to 24, 24 to 36, 36 to 48, and every 24 h out to 312 h. Samples were stored on ice until the end of the collection period. Aliquots were taken for radioanalysis and the urine samples were stored at approximately -20°C.
Fecal samples were collected in plastic bags and homogenized after the addition of water (if necessary to create a homogeneous slurry). Aliquots of each homogenate sample were combusted and then analyzed for trapped 14CO2. The remaining homogenized samples were stored in plastic containers at approximately -20°C.
A sample of expired breath was collected from each subject for analysis of 14CO2 within approximately 0.5 h before dosing (0-h sample), and at approximately 1, 2, 4, 6, 8, 10, 12, 16, 20, and 24 h after dosing. Subjects exhaled into a solution containing a trapping agent (benzethonium hydroxide) for the collection of expired CO2 and 14CO2.
Radioanalysis. Plasma, urine, and expired breath samples were prepared for analysis of radioactivity by adding Ready Protein+ (Beckman Coulter, Inc., Fullerton, CA) liquid scintillation cocktail to a known volume of sample. Radioactivity analysis was performed using a Packard Tri-Carb liquid scintillation spectrometer (model 2300; PerkinElmer Life Sciences, Boston, MA). Aliquots of the fecal homogenates were weighed into pretared combustion cones and allowed to dry overnight. The samples were combusted with a PerkinElmer Life Sciences Tri-Carb sample oxidizer (model 307) followed by liquid scintillation counting in Carbo-Sorb E/Permafluor E+ (PerkinElmer Life Sciences). Determination of [14C]duloxetine radioequivalents, data acquisition and storage, and statistical handling of the data were performed using the ADME/WINPET system (Eli Lilly and Co.) application and a validated Laboratory Information Management System (ADME/LIMS), based on an available software package (PerkinElmer Nelson's SQL*LIMS; PerkinElmer Instruments, Norwalk, CT). Subject number, sample weights or volumes, specific activity, and dpm results were used for the determination of percentage of dose excreted.
Analysis of Duloxetine. Plasma concentrations of the parent compound were determined using a validated LC/MS-MS assay. The assay was validated in the range of 0.5 to 100 ng/ml. Duloxetine and the stable isotope-labeled internal standard (13CD3 duloxetine HCl) were extracted from plasma by automated solid phase extraction using Ansys Spec Plus 96-well C8 extraction plates (Thames Restek UK Ltd., Windsor, UK) on a robotic liquid handling system (Multiprobe 204 Liquid Handling System, Canberra Industries, Pangborne, UK). The cartridges were conditioned with methanol followed by water prior to the addition of the plasma samples. The cartridges were then washed with water and 60% methanol in water (v/v). The samples were eluted with 100% methanol for direct injection by the autosampler. The compounds were separated chromatographically using an HI-RPB (30 mm × 3.2 mm i.d.) analytical column (Hichrom Ltd., Theale, Berkshire, UK) with mobile phase consisting of 55% acetonitrile/45% 10 mM ammonium acetate, pH 5 (v/v). The extracts were analyzed on a PerkinElmerSciex API III+ LC/MS-MS system using Turbo Ionspray (PerkinElmerSciex Instruments, Boston, MA). Tandem mass spectrometry was used to monitor the transition of m/z 298.1→44.0 for duloxetine and 302.1→48.0 for the internal standard. Standard curves and quality control samples were analyzed along with the samples. The overall percentage CV (precision) and percentage relative error (accuracy) of the assay after three validation runs were <8%. Duloxetine was stable in human plasma for at least 1 year when stored at approximately -20°C or -70°C.
Metabolite Profiling and Identification by HPLC. Plasma samples were pooled and extracted by precipitation with acetonitrile or by solid phase extraction (SPE) using a C8 cartridge (100 mg; Varian Medical Systems, Palo Alto, CA). A 1-ml plasma sample was precipitated by adding 3 ml of acetonitrile followed by centrifugation. The aqueous/organic layer was transferred to a silanized glass tube, evaporated under nitrogen, and reconstituted with 50% methanol/50% water (containing 0.1% trifluoroacetic acid) (v/v) and injected for analysis. The same reconstitution fluid was used with all of the plasma, urine, and fecal extractions. The extraction recovery of radioactivity was approximately 81%. For SPE, 1 ml of plasma was added to a preconditioned C8 SPE cartridge (1 ml of methanol followed by 1 ml of water) on a vacuum system. The plasma sample was slowly eluted through the cartridge and the cartridge was then washed with 1 ml of water. The metabolites were eluted with 2 ml of methanol into a silanized glass tube, evaporated under nitrogen, and reconstituted for injection. The extraction recovery of radioactivity was approximately 71%.
Urine samples were either directly injected (for those samples containing high amounts of radioactivity) or extracted using a C8 SPE cartridge prior to analysis. For SPE, 1 to 2 ml of urine was extracted using the same procedure as described for plasma. The extraction recovery of radioactivity was approximately 84%.
Plasma and urine samples were hydrolyzed by incubating a 1:1 mixture of sample and 0.2 M acetate buffer (pH 4.7) in a 37°C water bath overnight with β-glucuronidase containing sulfatase (type H-2 from Helix pomatia). The samples were then processed for analysis as described previously.
Fecal homogenates were extracted by adding approximately 5 ml of methanol to 1 to 2 g of sample. The sample was vortexed, mixed on a rotator for approximately 15 min, and centrifuged. The extract was transferred to a silanized tube, and the extraction procedure was repeated. The extracts were combined into one tube, evaporated under nitrogen, and reconstituted for injection. The extraction recovery of radioactivity was approximately 81%.
Duloxetine and its metabolites were separated on a Discovery C18 column (4.6 mm × 150 mm, 5 μm; Supelco, Bellefonte, PA) after passing through a 0.2-μm prefilter attached prior to the analytical column. A gradient separation was used to separate the metabolites. Mobile phase A consisted of 5% acetonitrile and 95% water (containing 0.1% trifluoroacetic acid) (v/v). Mobile phase B had an acetonitrile content of 75%. The gradient consisted of the following steps: 0 to 25 min, linear gradient from 100% A to 70% A; 25 to 35 min, isocratic at 70% A; 35 to 45 min, linear gradient to 0% A; 45 to 49 min, isocratic at 0% A; 49 to 50 min, linear gradient to 100% A; 50 to 60 min, isocratic at 100% A. HPLC flow was at 1 ml/min. Detection of the metabolites was by UV (232 nm) and radiodetection (IN/US β-RAM; IN/US Systems, Inc., Tampa, FL). The radiodetector utilized a 1,000-μl liquid cell with the flow rate of liquid scintillant (Ready Protein+) to mobile phase at a ratio of 4:1.
Due to the number of metabolites, HPLC mobile phase and gradient conditions were later improved to separate metabolites that coeluted in the urine samples. Mobile phase A consisted of 5% acetonitrile and 95% water (containing 0.025% trifluoroacetic acid and 0.075% formic acid, pH 2.5) (v/v). Mobile phase B had an acetonitrile content of 75%. The gradient consisted of the following steps: 0 to 12 min, linear gradient from 100% A to 85% A; 12 to 25 min, linear gradient to 80% A; 25 to 32 min, isocratic at 80% A; 32 to 35 min, linear gradient to 70% A; 35 to 40 min, isocratic at 70% A; 40 to 50 min, linear gradient to 0% A; 50 to 54 min, isocratic at 0% A; 54 to 55 min, linear gradient to 100% A; 55 to 65 min, isocratic at 100% A. HPLC flow was at 1 ml/min.
Metabolite Identification by Mass Spectrometry and NMR. The LC/MS analyses were performed using a Finnigan TSQ-7000 mass spectrometer (ThermoFinnigan, San Jose, CA). HPLC conditions (column and mobile phases) were identical to those used for metabolite profiling. However, different HPLC conditions were utilized for analysis of the sulfate metabolites. Mobile phase A consisted of 95% 10 mM ammonium acetate and 5% acetonitrile. Mobile phase B was 5% 10 mM ammonium acetate and 95% acetonitrile. The gradient consisted of the following steps: 0 to 5 min, hold at 100% A; 5 to 39 min, linear gradient to 20% A; 39 to 40 min, linear gradient to 0% A; 40 to 41 min, linear gradient to 100% A. HPLC flow rates for both conditions were at 1 ml/min. Approximately 25% of the eluant was diverted to the mass spectrometer via a 1:4 T splitter, and the remaining eluant (75%) was passed to a radiochemical detector (LB507; EG&G Berthold, Bad Wildbad, Germany). The radiodetector utilized a 500-μl liquid cell with a scintillant (Ultima Flo M; PerkinElmer Life Sciences) flow rate of 3 ml/min. This provided simultaneous detection of radioactivity and MS data. Mass spectral analysis was performed with both positive and negative ion electrospray ionization. The capillary temperature was 250°C. MS-MS analyses were performed using a collision energy of ±20 eV and a collision cell pressure using argon gas at 2.0 mTorr.
A Varian 400-MHz NMR instrument was used for synthesized standard structural characterization. Spectra were recorded at room temperature (locked at CD3OD signal) with Me4Si at 0 ppm as a reference. The metabolite structures were confirmed on either a Varian 600-MHz or 500-MHz NMR instrument. Either flow LC with column trapping or probe experiments were utilized for the various metabolites.
Pharmacokinetic Analysis. Plasma concentration versus time data for duloxetine and total radioactivity were analyzed using noncompartmental pharmacokinetic methods. Maximum observed plasma concentration and the corresponding sampling time were designated as Cmax and Tmax, respectively. Concentration-time data were plotted on a semilogarithmic scale, and the terminal log-linear phase was identified by visual inspection. The elimination rate constant (λz) was determined as the slope of the linear regression for the terminal log-linear portion of the concentration-time curve. A terminal half-life value (t1/2) was calculated as 0.693/λz. A predicted concentration (Č) at the last sampling time at which the assay value was above the limit of quantification was calculated from the regression equation. Area under the plasma concentration versus time curve (AUC0-t) was calculated by the trapezoidal method and extrapolated to infinite time as AUC0-∞ = AUC0-t + Č/λz.
Apparent plasma clearance (CLp/F) and volume of distribution (Vz/F) during the terminal elimination phase of duloxetine were calculated as CLp/F = Dose/AUC0-∞ and Vz/F = Dose/(λz × AUC0-∞), respectively.
Results
Clinical Signs. Duloxetine, administered orally as an enteric-coated 20.2-mg tablet, was well tolerated by all three of the male subjects in this study. The one female subject experienced nausea, retching (no actual emesis), headache, somnolence, and sweating within 3 h after duloxetine administration. Direct observation of this subject at the time of occurrence suggested that the phlebotomy procedure itself may have played a role in her symptoms. Regardless of their cause, these adverse events were mild to moderate and transient in nature. Electrocardiograms, laboratory reports, and vital signs showed no evidence of any safety problems related to duloxetine.
Excretion of Radioactivity in Urine and Feces. After oral administration of [14C]duloxetine in a 20.2-mg enteric-coated tablet, the total mean ± S.E.M. recovery of radioactivity at 312 h postdose was 90.5 ± 0.4% with 72.0 ± 1.1% excreted in urine and 18.5 ± 0.9% excreted in feces (Fig. 3). Radioactivity was not detected in the expired breath samples. The majority (87.2 ± 1.1%) of radioactivity was excreted within 120 h after dosing. The total recovery of radioactivity from individual subjects ranged from 89.5% to 91.1%.

Mean (±S.E.M.) cumulative elimination of radioactivity in urine and feces following a single oral 20.2-mg dose of [14C]duloxetine.
Pharmacokinetic Evaluation. The mean (SD) plasma concentration-time profiles of duloxetine and total radioactivity following oral administration of [14C]duloxetine in all four subjects are depicted in Fig. 4. Table 2 summarizes selected plasma pharmacokinetic parameters of duloxetine and total radioactivity in these individuals. Whereas the median Tmax was 6 h for both duloxetine and total radioactivity, the mean Cmax and AUC for duloxetine were approximately 9% and 3%, respectively, of the values for total radioactivity. The t1/2 of total radioactivity was 12-fold longer compared with that of duloxetine.
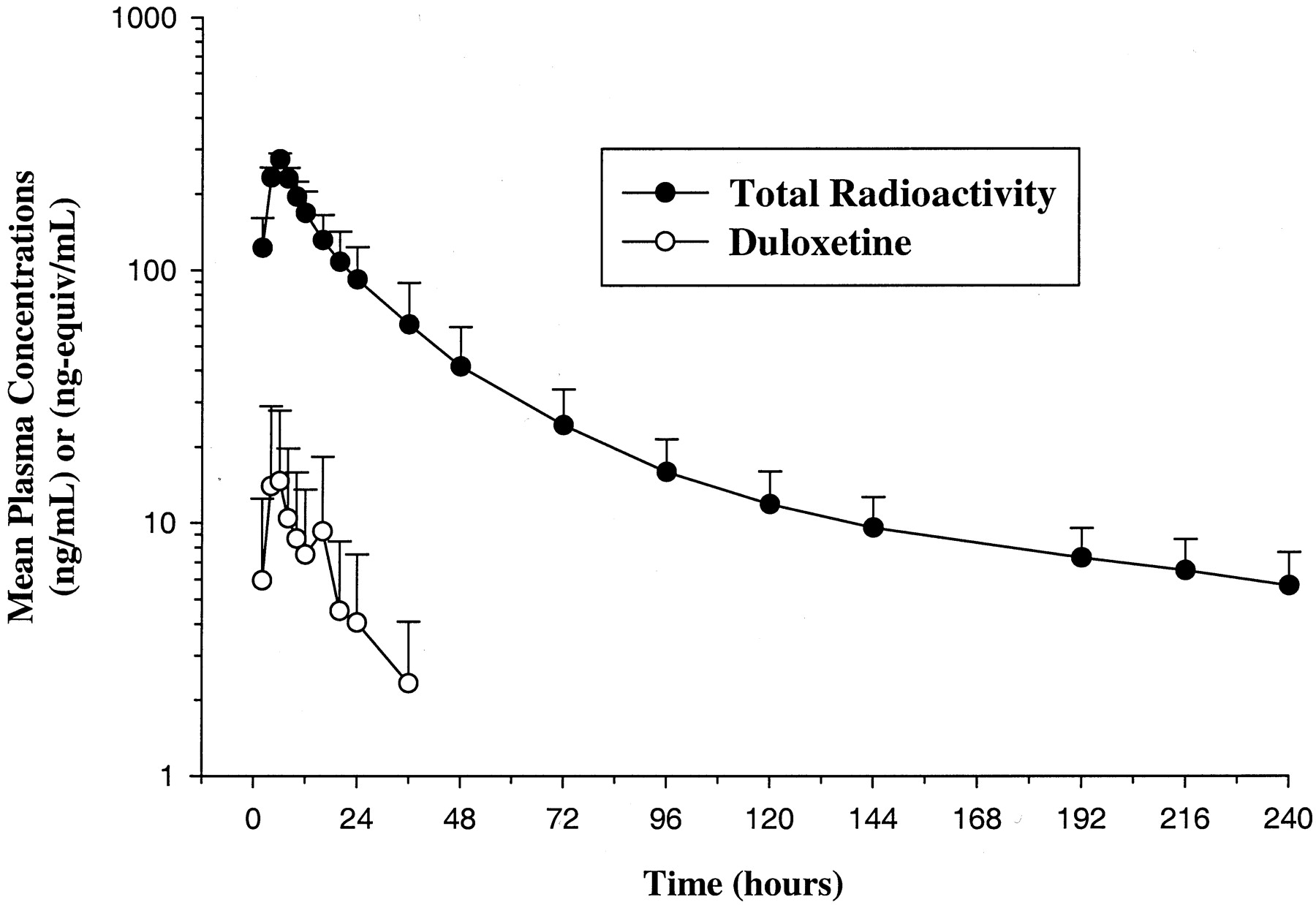
Mean (SD) duloxetine concentrations and total radioactivity in plasma following oral administration of [14C]duloxetine.
Pharmacokinetic parameters of duloxetine and total radioactivity in subjects receiving an oral dose of [14C]duloxetine
Metabolism. Plasma samples from each subject were pooled per sampling time point prior to extraction for metabolite analysis. Duloxetine, the glucuronide conjugate of 4-hydroxy duloxetine (M6), the sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7), the glucuronide conjugate of 4, 6-dihydroxy duloxetine (M9), and the glucuronide conjugate of 6-hydroxy-5-methoxy duloxetine (M10) were identified from the extracted plasma samples by comparison to synthetic standards and mass spectral data. The major metabolite in plasma was the glucuronide conjugate of 4-hydroxy duloxetine (M6). The second most abundant metabolite in plasma was the sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7). Radiochromatographic peak area percentages of duloxetine and the plasma metabolites are shown in Table 3. Figure 5 illustrates a radiochromatographic profile of plasma in addition to the profiles of urine and feces. Although only the 10-h plasma profile is shown in Fig. 5, the profiles of samples analyzed out to 48 h were similar.
Radiochromatographic HPLC peak area percentages of duloxetine and duloxetine metabolites in pooled plasma
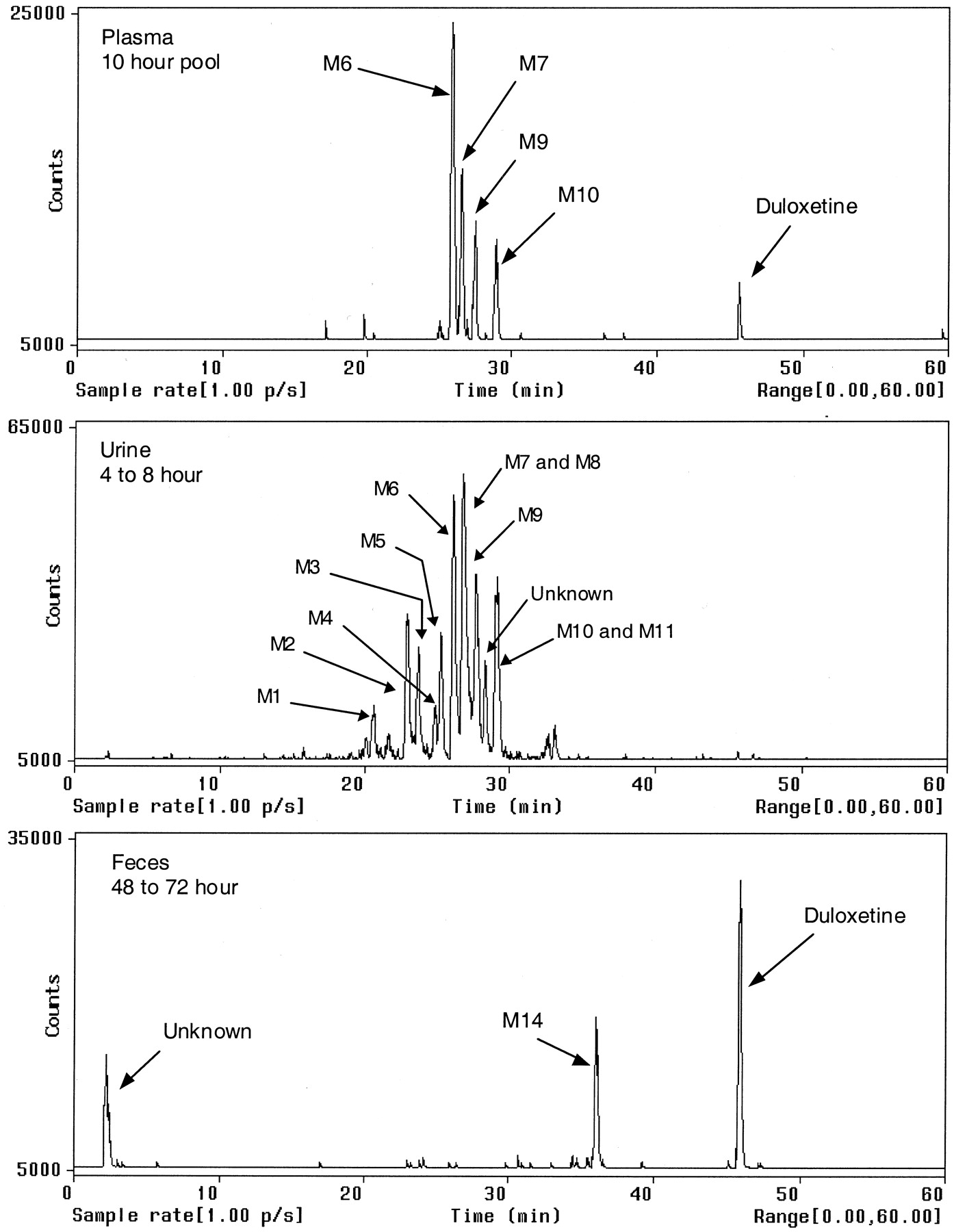
HPLC-radiochemical profiles for plasma, urine, and feces.
Urine samples were analyzed for metabolites only out to 72 h, since the levels of radioactivity in samples past 72 h were low and/or nondetectable. Approximately 94% (mean, n = 4) of the radioactivity excreted in the urine from each subject was eliminated by 72 h. Table 4 lists the individual and mean results of the percentage of total dose eliminated by 72 h for each metabolite based on radioactive peak areas. Results show that the glucuronide conjugate of 4-hydroxy duloxetine (M6) was the predominant metabolite, and it accounted for approximately 16.9% of the mean total dose. The sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7) was the second most abundant metabolite, and it accounted for approximately 12.5% of the mean total dose. Duloxetine was not detectable in the urine by HPLC/radiochemical detection.
Duloxetine metabolites in urine analyzed from 0 to 72 h expressed as individual and mean percentage of total dose
A fecal sample with the highest amount of radioactivity from each subject was extracted for metabolite analysis. The samples were either from the 48- to 72-h or 72- to 96-h collections. Two of three radioactive peaks were identified by mass spectrometry as duloxetine and 4-hydroxy duloxetine (M14), and one early eluting peak remains unknown. The radioactive peak area percentages of duloxetine and 4-hydroxy duloxetine were used to calculate the percentage of total dose for each sample (Table 5).
Percentage of total dose of duloxetine and its metabolites in feces
Metabolite Structure Elucidation by MS and NMR. Duloxetine was identified in plasma and feces. The retention time and product ion mass spectrum of the duloxetine peak matched that of the authentic duloxetine standard. The product ion mass spectrum of the protonated molecular ion m/z 298 produced the characteristic product ions at m/z 44 and m/z 154 corresponding to C2H6N and the loss of naphthol, respectively. These results confirmed the identification of the duloxetine peak in plasma and feces. The characteristic product ions of m/z 44 and m/z 154 were observed in the majority of the metabolites and were utilized as “characteristic markers” of duloxetine metabolites. In many instances these were the only fragment ions observed in the product ion mass spectrum along with an aglycone ion in some of the glucuronide and sulfate conjugates of duloxetine.
The glucuronide conjugate of 4-hydroxy duloxetine (M6) was found in plasma and urine producing a protonated molecular ion at m/z 490. This ion is 192 amu greater than that of duloxetine and indicates that M6 is a glucuronide conjugate of hydroxy duloxetine. The product ion mass spectrum of m/z 490 produced the product ions at m/z 44 and m/z 154. These ions suggest that the oxidation and subsequent glucuronidation of duloxetine has occurred on the naphthol ring. This peak was not present in the urine after hydrolysis with β-glucuronidase. Comparison of the retention time, product ion mass spectrum, and NMR spectrum with a synthesized standard of the glucuronide conjugate of 4-hydroxy duloxetine confirmed the identification of M6.
Metabolite M7 was found in plasma and urine. Both positive and negative electrospray ionization were utilized to determine the protonated and deprotonated molecular ions at m/z 424 and m/z 422, respectively. The characteristic ions for duloxetine at m/z 44 and m/z 154 were observed in the positive product ion mass spectrum of m/z 424. In addition, the product ions at m/z 344 and m/z 342 show the loss of 80 amu from m/z 424 and m/z 422, respectively. This loss is characteristic for sulfate conjugates. A product ion at m/z 314 indicates an addition of at least one oxygen moiety to duloxetine. However, this peak was present in urine after hydrolysis with β-glucuronidase. The synthesis of a standard and further detailed analysis with NMR confirmed the structural assignment of M7 as the sulfate conjugate of 5-hydroxy-6-methoxy duloxetine.
Metabolite M9 was also found in plasma and urine. The protonated molecular ion at m/z 506 did not produce the characteristic product ions of duloxetine at m/z 44 and m/z 154. However, the product ion at m/z 330 shows a loss of 176 amu from m/z 506, indicative of a glucuronide conjugate. This peak was not observed in urine following incubation with β-glucuronidase. A synthesized standard of 4,6-dihydroxy duloxetine was incubated with cell cultures to produce the glucuronide conjugates for comparison. Two different cell cultures (Streptomyces sp. and Actinoplanes missouriensis) produced the two glucuronide conjugates of 4,6-dihydroxy duloxetine. The M9 metabolite peak matched the retention time and product ion mass spectrum of a peak produced in the Streptomyces sp. cell culture incubation. This peak's structural assignment was determined with NMR analysis (Table 6). The proton chemical shifts for sites 2 and 3 on the naphthyl ring of the 4,6-dihydroxy duloxetine standard are magnetically equivalent and form a singlet at 6.63 ppm. In the case of metabolite M9, sites 2 and 3 are nonequivalent and form a doublet (6.99 ppm and 6.77 ppm), and have significantly shifted downfield (0.36 ppm and 0.14 ppm) compared with the 4,6-dihydroxy standard. In addition, the proton chemical shifts for sites 7 and 8 on the naphthyl ring for metabolite M9 (7.15 ppm and 8.15 ppm) and the 4,6-dihydroxy standard (7.13 ppm and 8.11 ppm) are insignificant (0.02 ppm and 0.04 ppm). The assignment of the structure of metabolite M9 was consistent with the observation that protons 2 and 3 shift downfield when the glucuronide residue was attached through the oxygen atom at position 4. Based on the MS and NMR data, metabolite M9 was confirmed as 4-O-glucuronide-6-hydroxy duloxetine (glucuronide conjugate of 4,6-dihydroxy duloxetine). M10 was found in plasma and urine and produced a protonated molecular ion at m/z 520. The characteristic product ions at m/z 44 and m/z 154 are observed. The product ion at m/z 344 shows a loss of 176 amu from m/z 520, indicative of a glucuronide conjugate. This peak was not found in urine after hydrolysis with β-glucuronidase and was converted to 6-hydroxy-5-methoxy duloxetine. The comparison of the retention time, product ion mass spectrum, and NMR analysis of M10 with that of a synthesized standard confirmed this metabolite as the glucuronide conjugate of 6-hydroxy-5-methoxy duloxetine.
1 H NMR data comparison of M9
Similar structural analyses were performed for M2, M3, M4, M8, M12 to M16, M23, and M26 and compared with synthesized standards. The retention time, product ion mass spectra, and NMR spectra were consistent with the structural assignments for these metabolites (Fig. 6). In addition, product ion mass spectra were obtained for M1 and M11. Both of these metabolites were not observed in urine after incubation with β-glucuronidase. Comparison of the retention time and product ion mass spectra with that of the other identified metabolites (and synthesized standards) produced the structural assignment of these two conjugates of duloxetine.
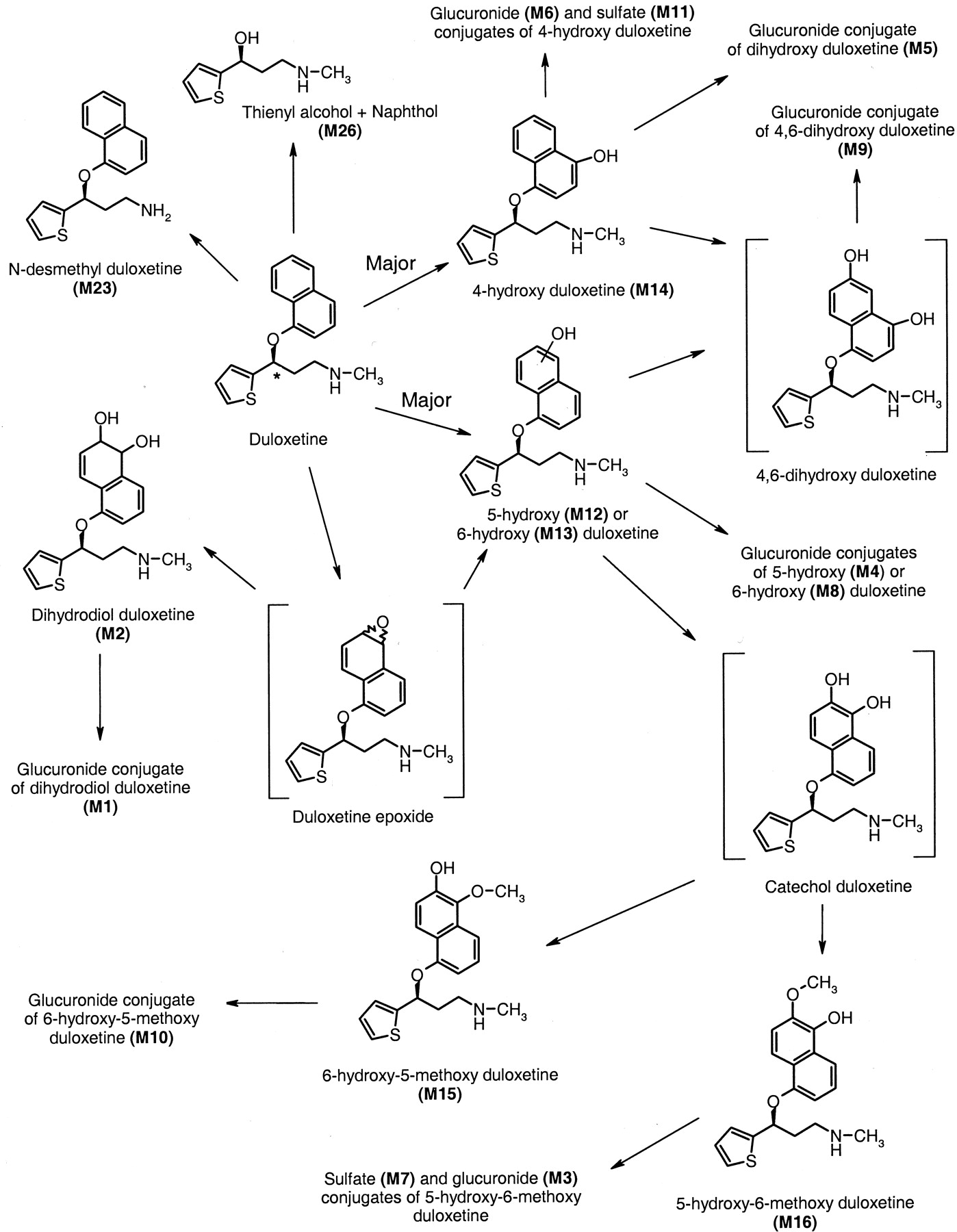
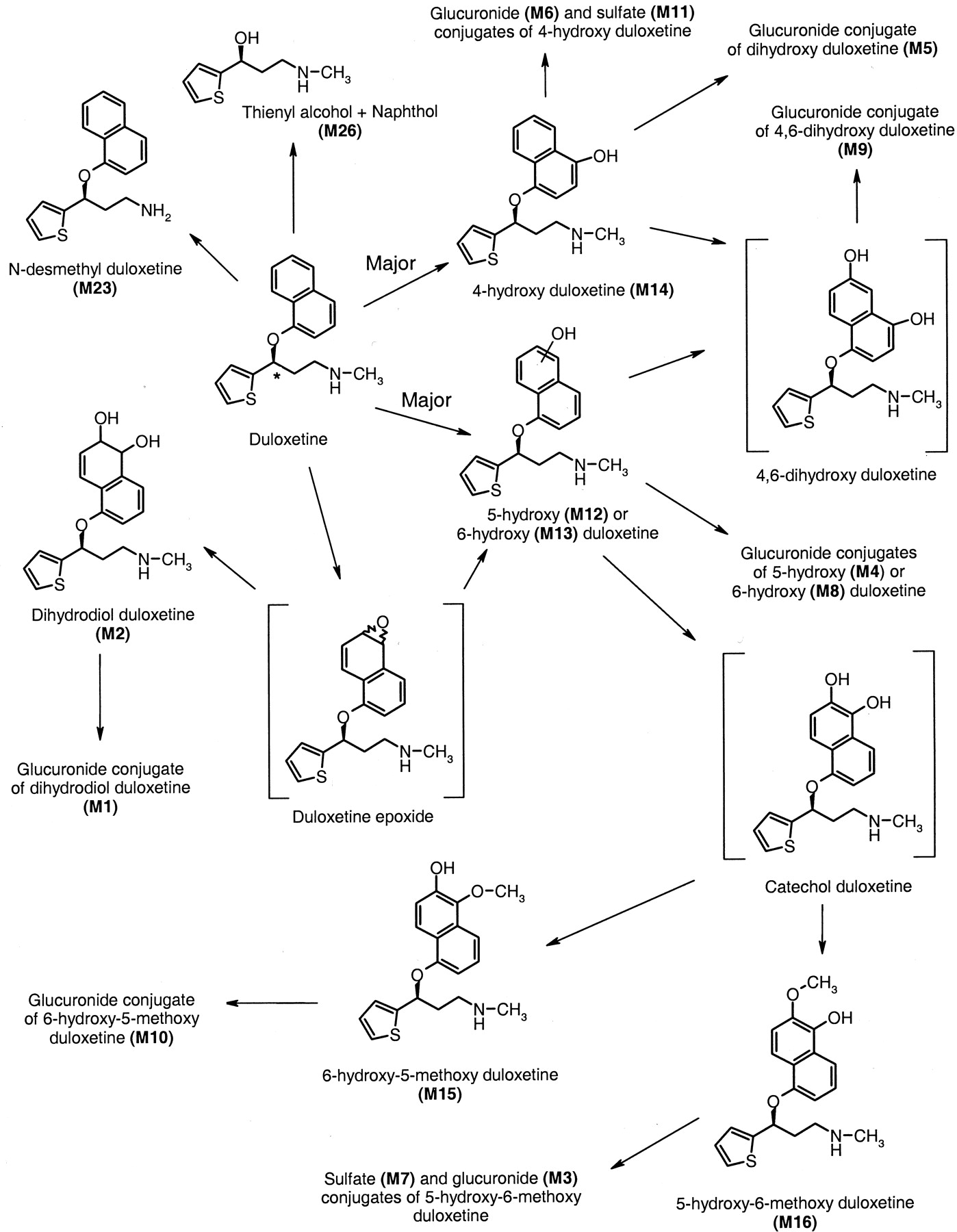
A schematic of the biotransformation pathways for duloxetine in humans.
The asterisk denotes the position of the 14C label on duloxetine.
Discussion
The biotransformation and disposition of duloxetine and its metabolites after administration of a single enteric-coated tablet containing 20.2 mg or 100.6 μCi of 14C-labeled duloxetine were characterized in this study. The mean total recovery of radioactivity was approximately 91%. The radioactivity was excreted primarily in urine (72%), with about 19% excreted in feces. The elimination pattern was similar between all the subjects. Figure 6 illustrates a schematic of the biotransformation pathways for duloxetine in humans.
Duloxetine was rapidly and extensively metabolized to form multiple oxidative and conjugated metabolites. Duloxetine accounted for only a small portion (approximately 3% for AUC and approximately 9% for Cmax) of the circulating radioactivity in plasma. The peak concentrations of total radioactivity occurred at the same time as that for duloxetine, with a Tmax of approximately 6 h. The late time of peak concentrations was presumably due to the enteric coating of the formulation used for duloxetine. In addition, the elimination half-life of total plasma radioactivity was much longer than that of duloxetine (120 h versus 10.3 h). These results suggest that the metabolism of duloxetine may occur on first-pass after oral administration and that radioactivity was eliminated more slowly than for duloxetine. The longer half-life of radioactivity could not be attributed to a single event (formation, distribution, or elimination) or to the elimination of a single metabolite. Duloxetine also appears to have a large apparent volume of distribution. The apparent plasma clearance, apparent volume of distribution, and half-life for duloxetine obtained in this study agree well with the previously published data on the pharmacokinetics of duloxetine (Sharma et al., 2000).
In plasma, the radioactivity was primarily glucuronide conjugates of 4-hydroxy duloxetine (M6), 6-hydroxy-5-methoxy duloxetine (M10), and 4,6-dihydroxy duloxetine (M9) or a sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7). The major metabolite in plasma was the glucuronide conjugate of 4-hydroxy duloxetine (M6). The unconjugated forms of 4-hydroxy duloxetine (M14), 6-hydroxy-5-methoxy duloxetine (M15), and 4,6-dihydroxy duloxetine were not detected in plasma using the radiodetection and mass spectrometry conditions used to identify the metabolites, although these compounds would be formed prior to conjugation. Plasma samples analyzed from other clinical studies in which duloxetine was dosed daily at 60 mg b.i.d. have shown a similar profile of circulating metabolites in plasma and confirm that the unconjugated forms are rapidly conjugated. Therefore, if these compounds circulate in plasma, they are very minor metabolites. The only difference between this radiolabeled study and the data at steady state was the presence of trace amounts of N-desmethyl duloxetine (M23) in the samples collected at steady state. The synthesized metabolites that were identified in both plasma and urine were tested for their affinity to human monoamine transporters and receptors, and they were found to be pharmacologically inactive (data on file).
The radioactivity was eliminated primarily in the urine within the first 96 h as glucuronide or sulfate conjugates of oxidative metabolites of duloxetine. This indicated that duloxetine was well absorbed, but extensively metabolized. The majority of the radioactivity found in feces was eliminated 48 h or more postdosing, suggesting that fecal radioactivity was probably due more to biliary excretion than to poor absorption of the compound. The profile of fecal radioactivity showed an unknown metabolite of duloxetine and 4-hydroxy duloxetine (M14) accounting for the majority of the fecal radioactivity.
All of the metabolites circulating in plasma were also detected in urine. Urine contained at least 11 different metabolites of duloxetine, with duloxetine itself being undetectable. All but one of these metabolites were glucuronide or sulfate conjugates. The major plasma metabolites were also the major urinary metabolites. Multiple biotransformation reactions occurred prior to elimination of the radioactivity. The initial biotransformation of duloxetine appears to be oxidation at either the 4-, 5-, or 6-position of the naphthyl ring. These hydroxyl compounds can then be conjugated or they can undergo further oxidation to form a catechol intermediate or another dihydroxy. The catechol can then undergo methylation to form a methyl catechol, which undergoes sulfation and glucuronidation. A minor pathway for 5- or 6-hydroxy duloxetine is the formation of a dihydrodiol, which is then glucuronidated. An intermediate in the formation of the dihydrodiol is an epoxide. The epoxide intermediate was chemically unstable and was short-lived since it was not detected in the samples, nor was a cysteine conjugate of the epoxide found in the samples. These data indicated the very rapid formation of the dihydrodiol from the postulated epoxide intermediate.
A very minor pathway was the cleavage of duloxetine at the chiral center to form a thienyl alcohol and naphthol. Since the molecule was labeled at the chiral center, once the cleavage occurred, the naphthol moiety would no longer contain the radiolabel and would not be detected. The thienyl alcohol (M26) and naphthol were detected in urine with the initial chromatographic/MS system at very low concentrations but were not detectable when the chromatographic and mass spectrometry conditions were optimized for metabolite separation and identification.
The clearance of duloxetine is mainly through the elimination of its metabolites. As stated previously, approximately 3% of the radioactivity in plasma was unchanged duloxetine. The main route of metabolism for duloxetine is the oxidation of the naphthol ring followed by conjugation. In vitro studies in human liver microsomes were performed to determine which human cytochrome P450 enzymes were responsible for the biotransformation of duloxetine to 4-, 5-, and 6-hydroxy duloxetine. Results of the studies have shown CYP2D6 and CYP1A2 to be the primary enzymes responsible for the oxidative metabolism at the 4-, 5-, or 6-position of the naphthyl ring (B. J. Ring et al., unpublished data). All four subjects in the study were found to be extensive metabolizers of CYP2D6. Further evidence of the involvement of CYP2D6 in the metabolism of duloxetine was obtained in a separate study in which coadministration of the potent CYP2D6 inhibitor, paroxetine, caused a 1.6-fold increase in the steady-state Cmax and AUC of duloxetine (Skinner et al., 2003). Differences in metabolism were not observed in smokers versus nonsmokers (two of four subjects smoked), although this study was too small to determine the effects of smoking on CYP1A2 expression.
In conclusion, duloxetine, administered orally as an enteric-coated 20.2-mg tablet, was shown to be safe and well tolerated by the subjects in this study. Duloxetine was extensively metabolized to numerous conjugative metabolites primarily excreted into the urine. Duloxetine accounted for a small percentage (<3%) of the circulating radioactivity based on mean AUC values.
Acknowledgments
We recognize and thank the following people whose work contributed to the manuscript. Steve Brooks analyzed plasma samples for duloxetine by LC/MS-MS and David Jackson helped with metabolite confirmation on the NMR. Milton Zmijewski prepared cell culture incubations for metabolite identification. We also thank Barb Ring, Jennifer Gillespie, John Bernstein, and Qimin Li for their work in determining which cytochrome P450 enzymes were responsible for duloxetine metabolism.
Footnotes
-
↵2 Abbreviations used are: 5HT, hydroxytryptamine (serotonin); NE, norepinephrine; LC/MS-MS, liquid chromatography/tandem mass spectrometry; HPLC, high performance liquid chromatography; SPE, solid phase extraction; AUC, area under the curve; amu, atomic mass unit(s).
-
↵1 Current address: Ligand Pharmaceuticals, 10275 Science Center Drive, San Diego, CA.
- Received March 10, 2003.
- Accepted June 6, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}