


Abstract
Ifosfamide nephrotoxicity is attributed to the formation of a toxic metabolite, chloroacetaldehyde, via N-dechloroethylation, a reaction that is purportedly catalyzed by CYP3A and CYP2B6. Because allelic variants of CYP3A5 are associated with polymorphic expression of microsomal CYP3A5 in human liver and kidneys, we hypothesized that ifosfamide N-dechloroethylation depends on CYP3A5 genotype. We compared ifosfamide N-dechloroethylation activity in cDNA-expressed CYP3A4 and CYP3A5. Ifosfamide N-dechloroethylation was also assessed in liver (N = 20) and kidney (N = 21) microsomes from human donors with different CYP3A5 genotypes. Ifosfamide N-dechloroethylation was catalyzed by recombinant CYP3A5 at a rate comparable with recombinant CYP3A4. In human liver microsomes matched for CYP3A4 protein content, N-dechloroethylation was more than 2-fold higher in that from donors carrying CYP3A5*1 allele that express CYP3A5 relative to that from donors homozygous for the mutant CYP3A5*3. Correlation analysis revealed that ifosfamide N-dechloroethylation was significantly associated with CYP3A4 and CYP3A5 protein concentration but not with age, sex, or CYP2B6 protein concentration. In hepatic microsomes not expressing CYP3A5 protein, ifosfamide N-dechloroethylation was inhibited 53 to 61% and 0 to 3% by monoclonal antibodies specific for CYP3A4/5 or CYP2B6, respectively. Ifosfamide N-dechloroethylation was not detected in renal microsomes obtained from CYP3A5*3/*3 donors. In contrast, it was readily measurable in microsomes isolated from four kidneys of CYP3A5*1 carriers, which was almost completely inhibited by the CYP3A inhibitor ketoconazole. CYP2B6 protein could not be detected in this panel of human renal microsomes. In conclusion, CYP3A5*1 genotype is associated with higher rates of ifosfamide N-dechloroethylation in human liver and kidneys.
The alkylating agent ifosfamide causes moderate-to-severe nephrotoxicity in 18 to 28% of patients (Loebstein et al., 1999; Skinner et al., 2000; McCune et al., 2004). Identifying patient-specific risk factors is of paramount importance to anticipate the potential for this long-term complication, because a chemoprotectant does not exist to prevent this adverse drug reaction. Cumulative ifosfamide dose greater than 60 g/m2 is the most commonly identified risk factor, but no safe dose limit exists (Loebstein et al., 1999; Skinner et al., 2000).
Ifosfamide itself is not cytotoxic in cultured renal epithelial cells or isolated perfused rat kidneys exposed to ifosfamide concentrations that have been observed in cancer patients receiving ifosfamide (i.e., 160–650 μM) (Kurowski and Wagner, 1993; Mohrmann et al., 1993; Zamlauski-Tucker et al., 1994; Springate et al., 1999; Boddy and Yule, 2000). This raises the possibility that ifosfamide metabolites are nephrotoxic. Ifosfamide is bioactivated via 4-hydroxylation to several reactive products, including acrolein. In parallel, ifosfamide is also metabolized by N-dechloroethylation to 2-dechloroethylifosfamide (2-DCEI), 3-dechloroethylifosfamide (3-DCEI), and an equivalent molar amount of chloroacetaldehyde. Both acrolein and chloroacetaldehyde cause toxicity in LLC-PK1 cells (Mohrmann et al., 1992, 1993); however, acrolein does not impair the function of isolated perfused rat kidneys or after long-term exposure in animal models (Parent et al., 1992; Zamlauski-Tucker et al., 1994). Chloroacetaldehyde has consistently shown a concentration-dependent cytotoxic effect in several in vitro models (i.e., porcine or rabbit cultured renal tubules and isolated perfused rat kidneys) with a minimum toxic concentration that ranges from 12.5 to 64 μM (Mohrmann et al., 1993; Springate, 1997; Springate et al., 1999). In cultured primary human proximal tubules cells, the minimum chloroacetaldehyde concentration for toxicity was reported to be 500 μM, which is well above the observed peak plasma concentrations (i.e., 0.5–109 μM) in patients receiving standard doses of ifosfamide (Goren et al., 1986; Kurowski and Wagner, 1993; Dubourg et al., 2001). Hence, circulating concentrations of chloroacetaldehyde is not sufficient to explain nephrotoxicity. It has recently been demonstrated that renal microsomes from pigs and humans catalyze ifosfamide N-dechloroethylation (Woodland et al., 2000; Aleska et al., 2001). Intrarenal ifosfamide N-dechloroethylation, with the release of proximate toxin chloroacetaldehyde, has been postulated to be the primary cause of renal toxicity (Aleksa et al., 2004).
In human liver microsomes, N-dechloroethylation of ifosfamide correlates well with the catalytic activities of CYP3A and CYP2B6 (Walker et al., 1994; Roy et al., 1999; Huang et al., 2000). A previous study by Gervot suggested renal expression of CYP2B6 protein, although the investigators cautioned that the observed immunoreactive protein may be CYP2B7 (Gervot et al., 1999). CYP3A4 and CYP3A5 are related isozymes that display 88% similarity in amino acid sequence and great overlap in their substrate specificity (Wrighton and Thummel, 2000). Expression of CYP3A4 is largely confined to the liver and intestinal mucosa; it has a negligible presence in the kidneys (Haehner et al., 1996; Koch et al., 2002; Givens et al., 2003). Tissue expression of CYP3A5 is more widespread and is a major P450 enzyme in the kidneys (Haehner et al., 1996; Wrighton and Thummel, 2000; Givens et al., 2003). However, it has been suggested that CYP3A5-mediated formation of chloroacetaldehyde from N-dechloroethylation of ifosfamide occurs at a much lower rate than that of CYP3A4 (Huang et al., 2000). This comparison was confounded by using two different expression systems of recombinant P450 (Supersomes): CYP3A4 coexpressed with cytochrome b5 versus CYP3A5 supplemented with an equimolar amount of exogenous cytochrome b5. It is known that catalytic activity of CYP3A4 is higher when coexpressed with cytochrome b5, in comparison with equimolar amounts of exogenous cytochrome b5 (Yamazaki et al., 1999). Perhaps the previously reported lower ifosfamide N-dechloroethylation by CYP3A5 simply reflected the difference in expression systems. Thus, the differences in rates of ifosfamide N-dechloroethylation between CYP3A4 and CYP3A5 should be verified in comparable expression systems.
Immunohistochemical studies have revealed localization of CYP3A5 in the epithelium of the proximal tubule, the thin loop of Henle, cortical collecting ducts, and the pelvis (Leggat et al., 1994). Renal CYP3A5 protein expression is polymorphic, resulting in profound differences (>100-fold) in CYP3A-dependent catalytic activity between individual donors (Haehner et al., 1996; Koch et al., 2002; Givens et al., 2003). There is strong evidence to suggest that inherited mutations in the CYP3A5 gene contribute to polymorphic protein expression/function. A single base substitution within intron-3 of CYP3A5 (6986A→G) creates an alternative acceptor splice site that results in the production of aberrant mRNA and a truncated nonfunctional protein (Kuehl et al., 2001). This mutation (CYP3A5*3) is the major cause of polymorphic CYP3A5 expression in the liver, small intestine, and kidneys (Lin et al., 2002; Givens et al., 2003). Thus, we hypothesize that rates of ifosfamide N-dechloroethylation in human liver and kidney are dependent on CYP3A5 genotype.
The specific aims of our studies were to 1) compare the rate of ifosfamide N-dechloroethylation by cDNA-expressed CYP3A4 and CYPA5 with equivalent cytochrome b5 supplementation or expression; 2) assess ifosfamide N-dechloroethylation activity in a panel of 20 liver microsomes matched for CYP3A4 content and in 21 kidney microsomes obtained from human donors with known CYP3A5 genotype; and 3) assess the relative contribution of CYP3A and CYP2B6 in ifosfamide N-dechloroethylation by microsomes isolated from human livers and kidneys.
Materials and Methods
Materials. NADPH, ketoconazole, thiotepa, and troleandomycin (TAO) were obtained from Sigma-Aldrich (St. Louis, MO). Ifosfamide was provided by the National Cancer Institute, Drug Synthesis and Chemistry Branch (Bethesda, MD). 2-Dechloroethylifosfamide and 3-dechloroethylifosfamide were kindly provided by Dr. Uif Niemeyer (Asta Medica, Frankfurt, Germany). Dipropylcyclophosphamide was synthesized as described by Kalhorn et al. (1999). Supersomes containing cDNA-expressed CYP3A4 + P450 reductase (lot no. 18, catalog 456207), CYP3A5 + P450 reductase (lot no. 18, catalog 456235), CYP3A4 + P450 reductase + cytochrome b5 (lot no. 55, catalog 456202), CYP3A5 + P450 reductase + cytochrome b5 (lot no. 1, catalog 456256), and CYP2B6 + P450 reductase + cytochrome b5 (lot no. 13513, catalog 456255) were purchased from BD Gentest (Woburn, MA). Cytochrome b5 (lot no. 23525) was purchased from PanVera Corp. (Madison, WI). Monoclonal antibodies directed toward CYP3A4/5 (catalog no. 458334) and toward CYP2B6 (catalog no. 458326) were also purchased from BD Gentest; mouse IgG (whole molecule) (catalog no. 015-000-003) was purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA) to serve as a control. Anti-human polyclonal antibody (catalog no. 458226) for Western blot of CYP2B6 within renal tissue was purchased from BD Gentest.
Tissue Collection and Preparation. Human livers were obtained from the University of Washington School of Pharmacy Tissue Bank (Seattle, WA). Liver microsomes were prepared by differential centrifugation and stored at –80°C as described previously (Lin et al., 2002). Twenty-one adult human kidneys were obtained from the National Disease Research Interchange (Philadelphia, PA); 17 of the donors were Caucasian, one was Hispanic, and three were of unknown ethnicity (Givens et al., 2003). Upon receiving the kidneys, microsomes were prepared by differential centrifugation and stored at –80°C as described previously (Dai et al., 2004). The final pellets were suspended in 100 mM potassium phosphate, 0.25 M sucrose, and 100 mM EDTA. Microsomal CYP3A5-specific content was measured by Western blot as described previously for liver (Lin et al., 2002) and renal microsomes (Dai et al., 2004), using a CYP3A5-specific antibody.
CYP3A5 Genotyping and Midazolam 1′-Hydroxylation. All procedures for CYP3A5 genotyping followed methods described previously for human liver (Lin et al., 2002). Single nucleotide polymorphisms in CYP3A5 corresponding to CYP3A5*3 (6986A→G), CYP3A5*6 (14690G→A), and CYP3A5*7 (27131–32, insertion T) allelic variants were evaluated. Midazolam 1′-hydroxylation activity of liver and renal microsomes used in this study has been reported previously (Lin et al., 2002; Givens et al., 2003).
CYP2B6 Protein Concentrations. CYP2B6 protein content of microsomal preparations from human kidneys was analyzed according to the method of Ekins et al. (1997). Microsomes (5–10 μg) were subjected to SDS-polyacrylamide gel electrophoresis; after SDS-polyacrylamide gel electrophoresis, proteins were transferred electrophoretically from the polyacrylamide gels to Immobilon P (Millipore Corporation, Billerica, MA) for immunoblotting using polyclonal antibodies produced against human CYP2B6-specific peptides.
IfosfamideN-Dechloroethylation by cDNA-Expressed CYP3A4 and CYP3A5. First, the saturation kinetics of ifosfamide N-dechloroethylation was evaluated using CYP3A4 + P450 reductase Supersomes and CYP3A5 + P450 reductase Supersomes; their respective cytochrome c reductase activity was 370 and 2200 nmol/(min × mg protein). Ifosfamide at concentrations ranging from 0.25 to 17.5 mM was preincubated with 10 pmol of recombinant enzyme and 30 pmol of cytochrome b5 (a 1:3 molar ratio) in a pH 7.4, 0.1 M potassium phosphate buffer containing 1 mM EDTA at 37°C for 5 min before addition of NADPH (1 mM final concentration). The incubation volume was 500 μl. After 15 min, the reactions were stopped with the addition of 4 ml of an ice-cold dichloromethane and isopropyl alcohol mixture (95%:5%). All incubations were performed in triplicate in silanized glass tubes to prevent adsorptive loss of 2-DCEI and 3-DCEI.
To evaluate the effect of cytochrome b5 supplementation, ifosfamide N-dechloroethylation by the two aforementioned Supersomes was measured with and without the addition of cytochrome b5 at an ifosfamide concentration of 0.25 mM. We also compared ifosfamide N-dechloroethylation between CYP3A4 + P450 reductase + cytochrome b5 Supersomes and the recently available CYP3A5 + P450 reductase + cytochrome b5 Supersomes, which are well matched in their activity of cytochrome c reductase (i.e., 5100 versus 3900 nmol/min/mg protein, respectively) and cytochrome b5 content (i.e., 1400 and 1600 pmol/mg protein, respectively).
Microsomal IfosfamideN-Dechloroethylation. Microsomal N-dechloroethylation activity was determined at a fixed ifosfamide concentration of 0.25 mM, which is well below the reported Km and within the plasma concentration range observed in patients (Kurowski and Wagner, 1993; Boddy and Yule, 2000). Initially, ifosfamide N-dechloroethylation was evaluated in a panel of 20 human liver microsomes, 10 of them from CYP3A5*3/*3 donors, and the remaining 10 from donors carrying a CYP3A5*1 allele. Each of the 10 pairs of microsomes was matched within 15% of CYP3A4 protein content. The levels of CYP2B6 in these human liver microsomes were determined by Western blot with polyclonal antibodies produced against human CYP2B6-specific peptides. Mean CYP2B6 protein concentrations did not differ between the two groups (p = 0.67). Ifosfamide at a final concentration of 0.25 mM was preincubated with 100 μg of human liver microsomal protein in 0.5-ml volume of phosphate buffer containing 0.1 mM EDTA. Incubations with liver microsomes were conducted in triplicate. After a 5-min preincubation, the reaction was initiated with the addition of NADPH at a final concentration of 1 mM. The incubations were stopped after 30 min by the addition of 4 ml of an ice-cold dichloromethane and isopropyl alcohol mixture (95%:5%).
Next, we evaluated ifosfamide N-dechloroethylation in 21 human kidney microsomes, four of which came from heterozygotes for the CYP3A5*1 allele, and the rest were from CYP3A*3/*3. Incubation conditions for the renal microsomes were identical to that for human liver microsomes, with the exception that the incubation duration was 120 min. Before initiating the human renal microsome experiments, the chemical stability of ifosfamide, 2-DCEI, and 3-DCEI over 120 min of incubation with boiled human liver microsomes was confirmed. We also assessed possible complication from secondary metabolism of 2-DCEI and 3-DCEI during a prolonged renal microsomal incubation. To conserve the limited amount of human kidney microsomes, we used a dilute preparation of human liver microsomes as controls. Based on the previously determined rates of midazolam 1′-hydroxylation in the human kidney and liver microsomes, we scaled down the protein content of one human liver preparation to simulate the expected activity of the human kidney microsomes. The ifosfamide N-dechloroethylation rate of this dilute liver microsomal preparation was later shown to be 500-fold higher than the average rate in CYP3A5*1/*3 human kidney microsome preparations. Using this dilute liver microsomal preparation, we confirmed that 2-DCEI and 3-DCEI concentrations were stable over 120 min, i.e., no product loss due to secondary metabolism. Control incubations with dilute human liver microsomes were included in each run: with ifosfamide and NADPH, NADPH alone (i.e., no ifosfamide), and ifosfamide alone (i.e., no NADPH). These controls provided a check for background, nonenzymatic formation of DCEIs.
Inhibition Studies. The quantitative contribution of CYP3A4 and CYP2B6 toward ifosfamide N-dechloroethylation in human liver microsomes was assessed by P450-specific chemical inhibitor and immunoinhibition studies. Similarly, chemical inhibition studies were conducted to assess the relative contribution of CYP3A5 and CYP2B6 in human renal microsomes. Specificity of the chemical inhibitors was checked using cDNA-expressed P450s, specifically, CYP2B6 + P450 reductase + cytochrome b5 with ketoconazole and TAO and CYP3A4 + P450 reductase + cytochrome b5 with thiotepa. The condition of immunoinhibition experiments with each monoclonal antibody were optimized using human liver microsomes based on manufacturers' specifications. Incubation conditions were identical to that described above, with the notable exceptions that the immunoinhibition experiments were conducted in plastic tubes and with 1 pmol of CYP3A4 + P450 reductase + cytochrome b5.
Preliminary experiments indicated that maximal inhibition of ifosfamide N-dechloroethylation was achieved in the presence of 10 μl of monoclonal antibody against CYP3A4/5 and CYP2B6; hence, this volume of monoclonal antibody was used in all subsequent experiments. For immunoinhibition, the monoclonal antibody and expressed enzymes or human liver microsomes were preincubated for 15 min on ice, followed by the addition of 37°C buffer. Ifosfamide was then added followed by an additional 5-min preincubation, and the reaction was started with the addition of NADPH. For both ketoconazole and thiotepa inhibition, ifosfamide and inhibitor were preincubated with expressed enzymes or human microsomes for 5-min, and reaction was started with the addition of NADPH. The final concentration of ketoconazole was 1 μM, which is 10 times its Ki for CYP3A5-mediated midazolam 1′-hydroxylation (109 ± 20 nM) (Gibbs et al., 1999). The final concentration of thiotepa was 50 μM, which is 10 times the Ki for CYP2B6-mediated S-mephenytoin N-demethylation in human liver microsomes (4.8 ± 0.3 μM) (Rae et al., 2002). TAO was preincubated with expressed enzyme or human liver microsomes, NADPH, and buffer for 30 min; the reaction was started with the addition of ifosfamide. The final TAO concentration was 10 μM, which has been reported to inhibit testosterone β-hydroxylation-CYP3A-catalyzed reaction by 85%, and did not inhibit bupropion hydroxylation, a CYP2B6-catalyzed reaction, in human liver microsomes (Faucette et al., 2001). All inhibition data are expressed relative to ifosfamide N-dechloroethylation in the same expression system or human microsomes without inhibitor conducted on the same day under the same conditions.
The proportion of ifosfamide N-dechloroethylation mediated by CYP3A4 and CYP2B6 was assessed in three human liver microsomal preparations that did not express CYP3A5 (i.e., CYP3A5*3/*3 genotype). The effects of ketoconazole and thiotepa were evaluated in human renal microsomes that contained CYP3A5 protein or those that produced 2-DCEI and 3-DCEI at concentrations greater than four times the limit of quantitation. Ketoconazole and TAO were used as CYP3A inhibitors (Gibbs et al., 1999; Faucette et al., 2001). Their potential overlap in inhibition toward CYP2B6 was checked using CYP2B6 + P450 reductase + cytochrome b5 Supersomes. Thiotepa was used as a specific CYP2B6 inhibitor (Rae et al., 2002). The effect of thiotepa on CYP3A-mediated ifosfamide N-dechloroethylation was checked using CYP3A4 + P450 reductase + cytochrome b5 Supersomes.
Analysis of Dechloroethylifosfamide Metabolites. Our early experience indicated that chloroacetaldehyde is inherently unstable in aqueous media, which poses tremendous difficulty in sample storage, handling, and analysis. Instead, we developed a sensitive and robust liquid chromatography-mass spectrometry method for quantitating the parallel by-products of ifosfamide N-dechloroethylation, 2-DCEI and 3-DCEI, in microsomal incubates. The sum of 2-DCEI and 3-DCEI formation was used as an index of chloroacetaldehyde production; the two products are formed at an equimolar ratio to chloroacetaldehyde (Norpoth, 1976). After termination of reactions, the internal standard (dipropylcyclophosphamide, 40 ng) was added to all samples and control incubates. The tubes were then capped and shaken on a reciprocating shaker at room temperature and then centrifuged for 10 min. The aqueous layer was removed from the samples, and the organic layer was transferred to silanized 12 × 75-mm glass culture tubes. The organic layer was evaporated to dryness at 40°C under a stream of dry air. The residue was redissolved in 50 μl of mobile phase and gently vortexed for 5 s. The resulting solution was transferred to an autosampler vial with insert and capped.
Calibration standards were prepared in inactivated (boiled) human liver microsomes. Standard curves were prepared in the range of 0.025 to 10 ng for each analyte; the limit of quantitation was 0.025 ng in a 0.5-ml sample. Nominal chromatographic separation was achieved on a 3-μm Hypersil BDS-C18 3.0 × 125 mm column heated to 35°C, using a mobile phase of acetonitrile in 20 mM ammonium acetate buffer. The acetonitrile gradient was set at 15% from time 0 to 3 min; 15 to 75% from 3 to 7 min, maintained at 75% from 7 to 15 min; and then decreased to 15% from 15 to 16 min. Retention times of 2-DCEI, 3-DCEI, and internal standard were 4.6, 5.4, and 10.5 min, respectively. The ionization mode was positive ion electrospray. Selective ion monitoring was set for m/z of 199 for 2-DCEI and 3-DCEI and 221.1 for the internal standard. Interday coefficient of variation for replicate analysis of quality control was 4.5% for 2-DCEI and 3.1% for 3-DCEI.
Data Analysis. Ifosfamide N-dechloroethylation rate versus substrate concentration data were fitted to a Michaelis-Menten model using the numerical module of the general modeling program SAAM (SAAM Institute, Seattle, WA). Statistical analysis was performed by SAS version 9.1 (SAS Institute, Cary, NC). Wilcoxon rank sum test was used for comparison of ifosfamide N-dechloroethylation by liver microsomes isolated from CYP3A5*3/*3 and *1/*3 donors. Multiple linear regression was used to estimate the association of ifosfamide N-dechloroethylation with the microsomal protein content of CYP3A4, CYP3A5, and CYP2B6 adjusted for the age and sex of the donor. The comparison of ifosfamide N-dechloroethylation rates in renal microsomes isolated from CYP3A5*3/*3 and *1/*3 donors was conducted using analysis of covariance. Detectable concentrations of 2-DCEI and 3-DCEI were observed in the 120-min control incubations (i.e., nonenzymatic N-dechloroethylation). To adjust for this background reaction, analysis of covariance was conducted with the dependent variable being the log of ifosfamide N-dechloroethylation rate and the independent variables being the CYP3A5 genotype and ifosfamide-NADPH control values. To assess the effects of ketoconazole and thiotepa inhibition, an analysis of variance was carried out treating kidneys as blocks. Post hoc comparisons of the ketoconazole and thiotepa groups with controls were carried out using Dunnett's test.
Results
We compared ifosfamide N-dechloroethylation by CYP3A4 + P450 reductase Supersomes and CYP3A5 + P450 reductase Supersomes supplemented with cytochrome b5 at a 1:3 molar ratio, and by Supersomes containing CYP3A4 coexpressed with P450 reductase and cytochrome b5 that was used in the previous study by Huang et al. (2000). The designated molar ratio of P450 to cytochrome b5 was set based on the study by Yamazaki et al. (1999), which observed maximal CYP3A4 activity at a 3-fold cytochrome b5 supplementation. As shown in Table 1, supplementation with cytochrome b5 at a 3-fold excess did lead to a modest increase in the rate of ifosfamide N-dechloroethylation for CYP3A5 but not for CYP3A4. CYP3A5 + P450 reductase Supersomes catalyzed ifosfamide N-dechloroethylation at a higher rate than CYP3A4 + P450 reductase Supersomes, either with or without cytochrome b5 supplementation. It seems that the previously observed higher catalytic rate of CYP3A4 coexpressed with cytochrome b5 relative to CYP3A5 with supplementation of cytochrome b5 reflected the difference between coexpression and supplementation of cytochrome b5 rather than an intrinsic difference in catalytic efficiency between CYP3A4 and CYP3A5 (Table 1). Our ifosfamide N-dechloroethylation rates were similar to chloroacetaldehyde formation rates reported by Huang et al. (2000) in CYP3A4 + P450 reductase + cytochrome b5 Supersomes (4.1 versus 5.43 pmol/min/pmol P450, respectively) and CYP3A5 + P450 reductase Supersomes supplemented with cytochrome b5 at a 1 to 1.5 ratio (0.330 versus 0.541 pmol/min/pmol P450, respectively). It should be noted that the two aforementioned Supersomes are not matched in terms of coexpressed P450 reductase activity, i.e., cytochrome c reductase activity of 2200 nmol/(min × mg protein) for CYP3A5 Supersomes compared with 370 nmol/(min × mg protein) for CYP3A4 Supersomes. Hence, the higher CYP3A5 Supersomes activity may be related to its higher coexpressed level of P450 reductase. An additional study with the recently available CYP3A5 + P450 reductase + cytochrome b5 Supersomes showed that ifosfamide N-dechloroethylation activity was 50% lower in CYP3A5 compared with CYP3A4 (Table 1). These Supersomes were well matched; coexpressed cytochrome c reductase activity at 3900 and 5100 nmol/(min × mg protein), and cytochrome b5 content was 1600 and 1400 pmol/mg protein for CYP3A5 and CYP3A4, respectively.
Effect of cytochrome b5 upon ifosfamide N-dechloroethylation (pmol of 2-DCEI and 3-DCEI formed/min/pmol P450) by cDNA-expressed CYP3A4 and CYP3A5
Data from the saturation experiments with CYP3A4 and CYP3A5, both supplemented with 1:3 molar ratio of cytochrome b5, are presented in Fig. 1. The Km and Vmax estimates for CYP3A4 were 2.63 mM and 3.11 pmol/min/pmol P450, respectively; intrinsic clearance (Vmax/Km) was 1.18 μl/min/nmol P450 (see Eadie-Hofstee plot in Fig. 2). For CYP3A5, the Km and Vmax were 1.55 mM and 5.48 pmol/min/pmol P450, respectively; intrinsic clearance was 3.54 μl/min/nmol P450. Thus, recombinant CYP3A5 has a slightly higher affinity for ifosfamide and is roughly 3-fold more efficient in mediating N-dechloroethylation of ifosfamide. The enzymes formed 2-DCEI and 3-DCEI at comparable amounts, i.e., similar regioselectivity.
As shown in Fig. 3 and Table 2, rate of ifosfamide N-dechloroethylation was more than 2-fold higher in liver microsomes from donors carrying the CYP3A5*1 allele that expressed CYP3A4 and CYP3A5 relative to microsomes from homozygotes of CYP3A5*3/*3 that expressed CYP3A4 only (p = 0.0001). Multiple regression analysis indicated that ifosfamide N-dechloroethylation was dependent upon CYP3A4 and CYP3A5 protein concentration (P = 0.0007 and 0.0230, respectively), but not upon age, sex, or CYP2B6 protein concentration (p > 0.05). For every 10-unit increase in CYP3A5 content (picomoles per milligram of protein), ifosfamide N-dechloroethylation would increase by 33 pmol/min/mg-protein, whereas the rate would be increased by only 8 pmol/min/mg-protein for every 10-unit increase in CYP3A4 content (picomoles per milligram of protein). These data suggest that CYP3A5 is the major microsomal N-dechloroethylase of ifosfamide in individuals carrying the CYP3A5*1 allele.
Ifosfamide N-dechloroethylation in liver and renal microsomes
Values are median (range) in pmol/min/mg protein.
The contribution of CYP3A4 and CYP2B6 was quantified by isoform-specific inhibitor studies with three human liver microsomal preparations that did not express CYP3A5 (i.e., CYP3A5*3/*3 genotype). The three microsomes were chosen from approximately 30 of the available human liver microsomes at the University of Washington Tissue Bank to represent varying proportions of the two enzymes: low CYP3A4 protein + high CYP2B6 protein, moderate expression of both enzymes, and high CYP3A4 protein + low CYP2B6 protein. Addition of ketoconazole (1 μM), a recognized CYP3A inhibitor, led to a similar degree of inhibition (i.e., 69–81%) regardless of the CYP3A4 protein concentration; a similar trend was noted with 10 μM TAO, which inhibited ifosfamide N-dechloroethylation within 5% of that of ketoconazole (Table 3). The CYP3A4/5 monoclonal antibody inhibited microsomal ifosfamide N-dechloroethylation to a lesser degree (53–61%), suggesting that ketoconazole and TAO might not be specific for CYP3A4. Indeed, ketoconazole and TAO inhibited ifosfamide N-dechloroethylation in cDNA-expressed CYP2B6 by 40 and 60%, respectively. The degree of inhibition by 50 μM thiotepa, a CYP2B6 inhibitor, did increase with an increasing amount of CYP2B6 protein (Table 3). However, this concentration of thiotepa inhibited ifosfamide N-dechloroethylation by 63% in CYP3A4 + P450 reductase + cytochrome b5 Supersomes. In contrast, CYP2B6 monoclonal antibody experiments demonstrated a negligible (0–3%) inhibition of ifosfamide N-dechloroethylation in human liver microsomes.
Inhibition of ifosfamide N-dechloroethylation in human liver microsomes isolated from CYP3A5*3/*3 donors
All donors were women between 45 and 60 years of age.
Ifosfamide N-dechloroethylation could not be detected in human renal microsomes that were homozygotes for CYP3A5*3/*3. Significant but variable rates of N-dechloroethylation were observed in heterozygotes of CYP3A5*1, suggesting that CYP3A5 is a critical determinant of chloroacetaldehyde formation in the human kidneys (Fig. 4). The median ifosfamide N-dechloroethylation rate in the four kidneys obtained from CYP3A5*1 carriers was 0.26 pmol/min/mgprotein, which is comparable with that reported by Woodland et al. (2000) in renal microsomes from one human subject (i.e., 0.19 pmol/min/mg-protein). Ketoconazole (1 μM) inhibited ifosfamide N-dechloroethylation by 93–100% in these four kidneys (P < 0.05), relative to control incubations without ketoconazole (Fig. 4). Thiotepa (50 μM) inhibited ifosfamide N-dechloroethylation by 12 to 31% in these four kidneys (P > 0.05) relative to control incubations without thiotepa (Fig. 4). We also checked for CYP2B6 protein expression in human kidney microsomes, which was below detection limit (1 pmol/mg protein) in seven human kidney microsomes.

Saturation kinetics of ifosfamide N-dechloroethylation.

Eadie-Hofstee plot of ifosfamide N-dechloroethylation by CYP3A4 and CYP3A5.
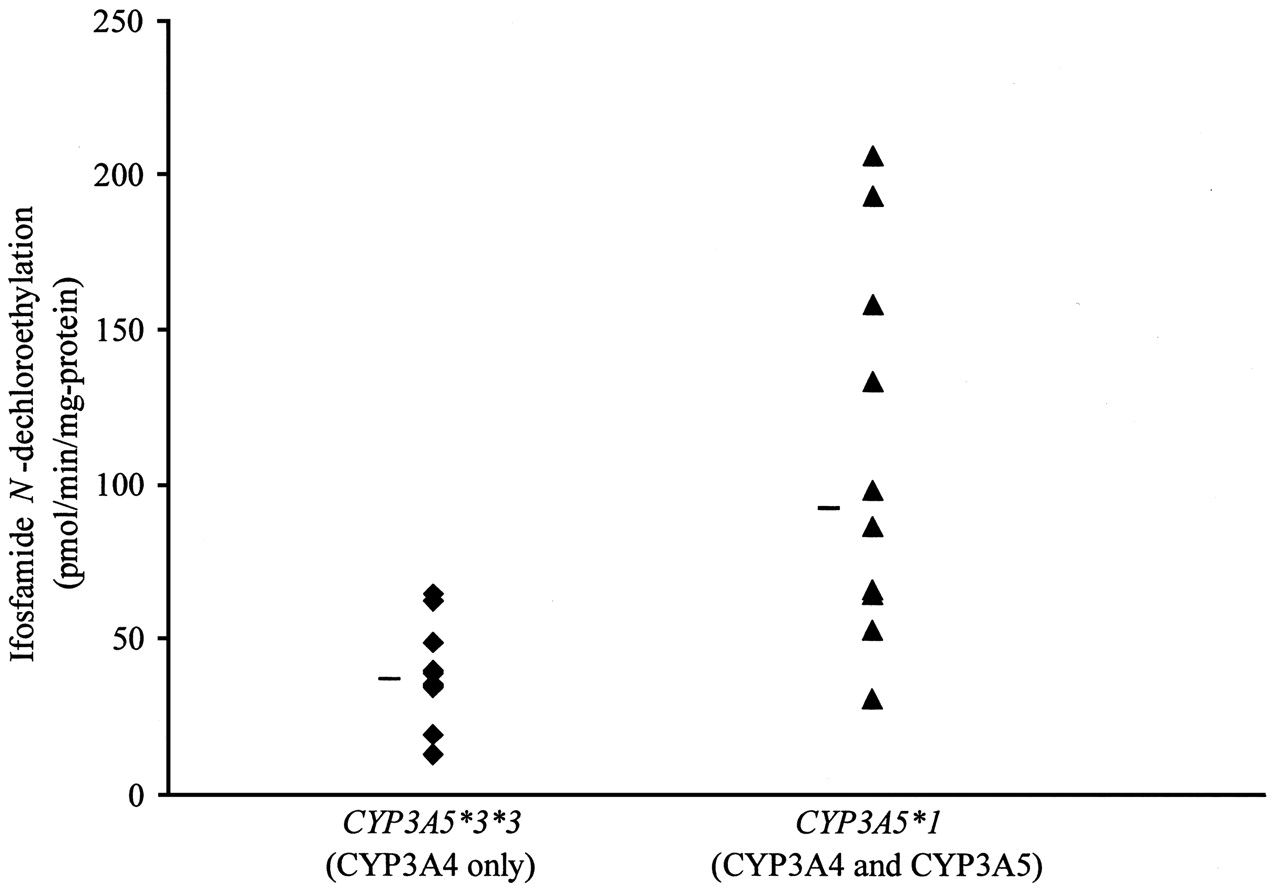
Variation in ifosfamide N-dechloroethylation rate in human liver microsomes as a function of CYP3A5 genotype. Hatch marks represent the median of that group.
Discussion
CYP3A5-catalyzed ifosfamide N-dechloroethylation is comparable or more efficient than CYP3A4 when the measurements were conducted using the same cytochrome P450 expression systems, at the same level of cytochrome b5 supplementation or coexpression (Table 1). In the presence of fortified cytochrome b5, the intrinsic clearance of ifosfamide N-dechloroethylation by CYP3A5 was 3-fold greater than that for CYP3A4 (i.e., 3.54 versus 1.18 μl/min/nmol P450). We also observed that cytochrome b5 supplementation increased the velocity of the N-dechloroethylation reactions by CYP3A5 but not CYP3A4; Yamazaki et al. (1999) and Dai et al. (2004) previously noted this trend with several other CYP3A substrates. Moreover, we were able to reproduce the apparent difference in ifosfamide N-dechloroethylation rate between the CYP3A4 and cytochrome b5 coexpression system and CYP3A5 with a 1:1.5 molar ratio cytochrome b5 supplementation as reported by Huang et al. (2000). It now seems likely that the previously reported higher rate of ifosfamide N-dechloroethylation in CYP3A4 compared with CYP3A5 by these investigators was confounded by differences in the two expression systems, i.e., coexpression versus supplementation of cytochrome b5. It should, however, be noted that the content of P450 reductase does differ between the Supersome preparations of CYP3A4 and CYP3A5, i.e., 370 versus 2200 nmol/(min × mg protein), respectively. Hence, we cannot rule out that the higher CYP3A5 activity could in part be due to the higher reductase expression. Subsequent experiments with the recently available Supersomes CYP3A5 coexpressed with P450 reductase and cytochrome b5 showed that this enzyme catalyzes ifosfamide N-dechloroethylation at approximately one-half the rate of CYP3A4 in a similar expression system that was well matched for cytochrome c and cytochrome b5 (Table 1).
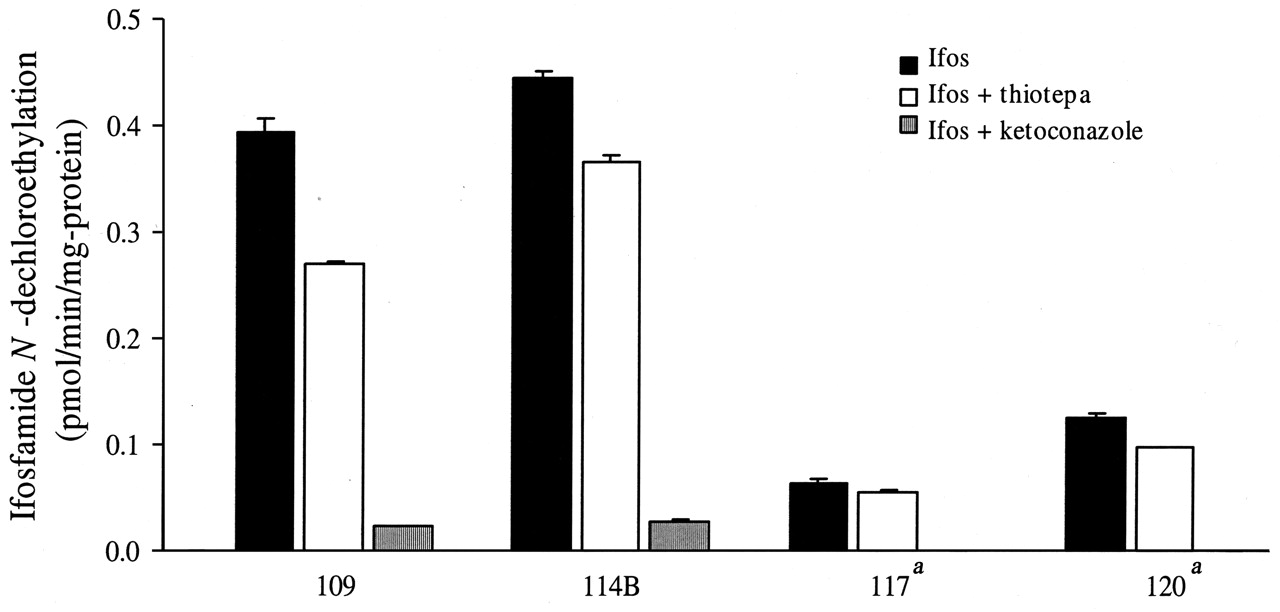
Effect of thiotepa and ketoconazole upon ifosfamide N-dechloroethylation in human kidney microsomes from CYP3A5*1 carriers. Data shown as means with standard deviation. a No detectable N-dechloroethylation of ifosfamide in the presence of ketoconazole.
We also showed in a panel of human liver microsomes that ifosfamide N-dechloroethylation is highly correlated with CYP3A4 and CYP3A5 protein content but not with CYP2B6 protein content, age, and gender of the donor. In pairs of human liver microsomes matched for their CYP3A4 protein content, N-dechloroethylation of ifosfamide was approximately 2-fold higher in liver microsomes obtained from donors carrying a CYP3A5*1 allele than in those from CYP3A5*3/*3 donors (Fig. 3). Our present comparative activity data between CYP3A5 genotypes for ifosfamide N-dechloroethylation agree with other CYP3A-dependent reactions reported previously with the same matched set of microsomes (e.g., midazolam 1′-hydroxylation and erythromycin N-demethylation) (Lin et al., 2002; Huang et al., 2004). Thus, ifosfamide seems to be another addition to the growing list of drugs for which hepatic CYP3A5 contribute significantly to their oxidative metabolism in individuals carrying CYP3A5*1 (Huang et al., 2004).
The relative contribution of CYP3A4 and CYP2B6 in ifosfamide N-dechloroethylation was examined by selective inhibitors in three human liver microsomes with varying CYP3A4 and CYP2B6 content (Table 3). The microsomes used in these experiments were from CYP3A5*3*3 donors to minimize any confounding effects of CYP3A5 because an inhibitor completely specific for CYP3A4 has not yet been reported. Ketoconazole and thiotepa were initially evaluated; however, recent data from Turpeinen et al. (2004) demonstrated that ketoconazole inhibits CYP2B6-catalyzed bupropion hydroxylation with an IC50 of 3.5 μM, which is slightly above the 1 μM concentration used in these experiments. It has been reported that 10 μM TAO does not inhibit bupropion hydroxylation, a CYP2B6-catalyzed reaction, in pooled human liver microsomes (Faucette et al., 2001). However, in this study both ketoconazole and TAO inhibited ifosfamide N-dechloroethylation by cDNA-expressed CYP2B6 + P450 reductase + cytochrome b5 to the extent of 40 and 60%, respectively. This explains the higher percentage of inhibition of N-dechloroethylation in human liver microsomes by both ketoconazole and TAO (65–81%) compared with anti-CYP3A4/5 monoclonal antibody (53–61%). Inhibition of N-dechloroethylation by thiotepa also lacked specificity; thiotepa inhibited ifosfamide N-dechloroethylation by 63% in CYP3A4 + P450 reductase + cytochrome b5 Supersomes. Our result stands in contrast to the report of Rae et al. (2002), who observed that thiotepa had minimal effect upon CYP3A activity (i.e., midazolam 4′-hydroxylation). CYP2B6 immunoinhibition experiments demonstrated barely measurable (<3%) inhibition of ifosfamide N-dechloroethylation in human liver microsomes. Thus, these data suggest that CYP3A4 plays a predominant role in hepatic ifosfamide N-dechloroethylation, whereas CYP2B6 has a negligible role, even in those hepatic microsomes with a high expression of CYP2B6 protein (i.e., HLM 142; Table 3). Our data contradict those reported by Huang et al. (2000), who reported that CYP2B6 monoclonal antibody inhibited ifosfamide N-dechloroethylation by 40% in human liver microsomes. In another study by Granvil et al. (1999), CYP2B6 activity (i.e., S-mephenytoin N-demethylation) was highly correlated (R2 > 0.95) with the formation rate of S-3-dechloroethylifosfamide and S-2-dechloroethylifosfamide from (R)-ifosfamide and (S)-ifosfamide, respectively; however, the relative contribution of CYP3A4/5 and CYP2B6 toward N-dechloroethylation of each ifosfamide enantiomer was not evaluated (Granvil et al., 1999). We have no obvious explanation for the discrepancy between our data and the previous study by Huang et al. (2000). Further studies are needed regarding the relative importance of CYP3A4/5 and CYP2B6 in hepatic ifosfamide N-dechloroethylation.
Until recently, it had been assumed that hepatic ifosfamide N-dechloroethylation leads to the release of chloroacetaldehyde into the systemic circulation and would inflict nephrotoxicity upon reaching the kidneys. However, this scenario is questionable because peak plasma chloroacetaldehyde concentrations range from 0.5 to 109 μM in patients receiving standard doses of ifosfamide, which are appreciably lower than its reported minimum nephrotoxic concentration (Goren et al., 1986; Kurowski and Wagner, 1993; Dubourg et al., 2001). In freshly isolated human renal proximal tubules, the minimal chloroacetaldehyde concentration leading to toxicity (e.g., lactate dehydrogenase release and impaired lactate metabolism) was 500 μM (Dubourg et al., 2001). This has led to the alternative hypothesis that ifosfamide N-dechloroethylation is mediated by CYP3A within renal proximal tubule, i.e., chloroacetaldehyde is produced locally at the site of toxicity (Woodland et al., 2000; Aleska et al., 2001, 2004).
Ifosfamide N-dechloroethylation could not be detected in human renal microsomes from CYP3A5*3*3 donors, i.e., when CYP3A5 expression is negligible. In the renal microsomes obtained from CYP3A5*1 carriers, ifosfamide N-dechloroethylation ranged from 0.06 to 0.45 pmol/min/mg-protein (Table 2; Fig. 4). This catalytic activity was effectively inhibited by ketoconazole but not by thiotepa (Fig. 4). Although the presence of CYP2B6 in human renal tubules has been suggested (Gervot et al., 1999; Aleksa et al., 2004), we could not detect CYP2B6 protein in our panel of human renal microsomes. Thus, current data suggest that CYP2B6 does not seem to be important in renal ifosfamide N-dechloroethylation.
These data predict that cancer patients who carry CYP3A5*1 would catalyze ifosfamide N-dechloroethylation at a greater rate, leading to higher chloroacetaldehyde concentrations and a higher risk of ifosfamide-induced nephrotoxicity. The effects of renal ifosfamide N-dechloroethylation are expected to be the primary contributor to nephrotoxicity because chloroacetaldehyde degrades rapidly in human blood (Kurowski and Wagner, 1993). Interestingly, there is increasing recognition for the importance of extrahepatic CYP3A5 in the pathophysiology of hypertension and prostate cancer risk (Givens et al., 2003). Although mesna and amifostine are capable of preventing chloroacetaldehyde-induced renal toxicity in vitro, they have not proven to be clinically effective (de Kraker et al., 2000; Zaki et al., 2003). Therefore, efforts to minimize ifosfamide nephrotoxicity have focused upon identifying patient-specific risk factors (Loebstein et al., 1999; Skinner et al., 2000; McCune et al., 2004). Further studies are now needed to demonstrate the association of CYP3A5*1 with ifosfamide nephrotoxicity in cancer survivors.
Footnotes
-
This study was supported by University of Washington Royalty Research Fund Award #2578.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002279.
-
ABBREVIATIONS: 2-DCEI, 2-dechloroethylifosfamide; 3-DCEI, 3-dechloroethylifosfamide; P450, cytochrome P450; TAO, troleandomycin.
- Received September 13, 2004.
- Accepted April 6, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}