


Abstract
Members of the human CYP3A family of metabolizing enzymes exhibit developmental changes in expression whereby CYP3A7 is expressed in fetal tissues, followed by a transition to expression of CYP3A4 in the first months of life. Despite knowledge about the general pattern of CYP3A activity in human development, the mechanisms that regulate developmental expression remain poorly understood. Epigenetic changes, including cytosine methylation, have been suggested to play a role in the regulation of CYP3A expression. The objective of this study was to investigate changes in cytosine methylation of the CYP3A4 and CYP3A7 genes in human pediatric and prenatal livers. The methylation status of cytosine-phospho-guanine dinucleotides was determined in 16 pediatric liver samples using methyl-seq and confirmed by bisulfite sequencing of 48 pediatric and 34 prenatal liver samples. Samples were separated by age into five groups (prenatal, < 1 year of age, 1.8–6 years, 7–11 years, and 12–17 years). Methyl-seq anaylsis revealed that cytosines in the proximal promoter of CYP3A7 are hypomethylated in neonates compared with adolescents (P < 0.001). In contrast, a cytosine 383 base pair upstream of CYP3A4 is hypermethylated in liver samples from neonates compared with adolescents (P = 0.00001). Developmental changes in methylation of cytosines in the proximal promoters of CYP3A4 and CYP3A7 in pediatric livers were confirmed by bisulfite sequencing. In addition, the methylation status of cytosine in the CYP3A4 and CYP3A7 proximal promoters correlated with changes in developmental expression of mRNA for the two enzymes.
Introduction
Developmental changes in the expression of members of the human cytochrome P450 3A family have been well documented whereby CYP3A7 is expressed at high levels in prenatal and neonatal livers, followed by a transition to CYP3A4 expression in the first months of life (Lacroix et al., 1997; Stevens et al., 2003). Despite an understanding about changes in the general pattern of CYP3A expression and activity in humans during development, the mechanisms that regulate changes in developmental expression remain poorly understood. In addition to developmental changes in expression, members of the CYP3A family exhibit extensive interindividual variability in expression and activity, which may be due to genetic and environmental factors. In terms of genetic variation, the CYP3A5*3 allele is largely responsible for the polymorphic expression of the enzyme (Kuehl et al., 2001), and the CYP3A7*1C and CYP3A7*1B alleles have been associated with sustained expression of CYP3A7 in adult tissues (Burk et al., 2002; Sim et al., 2005). The relative contribution of CYP3A5 or CYP3A7 to total CYP3A activity in adults remains a matter of debate, however, and is likely substrate-dependent.
In contrast, polymorphisms within the CYP3A4 gene do not adequately explain the observed variability in expression. Clinically, only CYP3A4*22 has consistently been associated with reduced catalytic activity (Elens et al., 2011; Wang et al., 2011), however, it is unlikely to be a major determinant of CYP3A4 activity in the broader context, with a minor allele frequency of just 3%–8% in the white population. The lack of commonly occurring genetic variation in the CYP3A4 gene has led to the hypothesis that variability in factors responsible for regulating constitutive and inducible expression of CYP3A4 is responsible for interindividual variability (Schuetz, 2004). Polymorphisms in liver-enriched transcription factors, including FOXA2 (HNF3β), HNF4A, FOXA3 (HNF3γ), NR1I2 (pregnane, PXR), ARNT, GR, PGRMC2, and PPARA, as well as polymorphisms in transporters (e.g., ABCB1, SLCO1B3), have been investigated for their effects on CYP3A4 expression and activity (Franke et al., 2008; Lamba et al., 2010; Klein et al., 2012). Using these approaches, Klein et al. (2012) were able to account for 33% of the variability in 2-OH-atorvastatin formation in vivo when genetic and nongenetic (e.g., gender) factors were combined.
Epigenetic changes, including cytosine methylation, have been suggested to play a role in the regulation of CYP3A expression (Medina et al., 2005; Li et al., 2009; Kacevska et al., 2012). In cultured hepatoma (HepG2) and colon cancer (Caco-2) cell lines treated with 5-aza-2′-deoxycytidine (5-aza-dC), mRNA expression of members of the CYP3A family increased over baseline via direct changes in DNA methylation of the CYP3A genes or indirectly via altered methylation of the gene encoding PXR (Dannenberg and Edenberg, 2006; Habano et al., 2011). In human liver tissue, variability in CYP3A4 mRNA expression was correlated with the methylation state of cytosines in the proximal promoter at position −1547 base pairs (bp, relative to the ATG translation start codon) and near the constitutive liver enhancer module of CYP3A enhancer region at position −10,762 bp (Kacevska et al., 2012). In addition to associations with interindividual variability in CYP3A expression, data from mice also indicate that DNA methylation of the Cyp3a locus in association with alterations in histone methylation patterns contribute to the developmental switch between Cyp3a11 and Cyp3a16 expression in mouse postnatal livers (Li et al., 2009).
In general, hypermethylation of cytosine in cytosine-phospho-guanine (CpG) dinucleotides near the 5′ region of a gene is associated with silencing of gene expression, whereas hypomethylation is associated with activation of gene expression (Brenet et al., 2011). Although no CpG islands are found within the human CYP3A locus [CYP3A CpGobs/CpGexp = 0.157 across 220 kilobase (kb) versus CpG islands CpGobs/CpGexp > 0.6], data now suggest that methylation not only in CpG islands but also in CpG shores/shelves and even individual CpG dinucleotides may play a role in the regulation of gene expression (Nile et al., 2008; Claus et al., 2012). Therefore, the objective of this study was to investigate changes in CpG methylation of the proximal promoters of the CYP3A4 and CYP3A7 genes in prenatal and pediatric human livers. DNA methylation levels in the proximal promoters of CYP3A4 and CYP3A7 were then compared with gene expression levels in the same tissues to investigate the potential interaction between DNA methylation and developmental changes in expression in human livers.
Materials and Methods
Pediatric Liver Samples.
Forty-eight anonymized pediatric liver samples, from children aged 0 days to 17 years (10 girls, 38 boys), were received from the following sources: the National Institute of Child Health and Human Development Brain and Tissue Bank at the University of Maryland [funded by National Institutes of Health (NIH) contract HHSN275200900011C, ref. no. N01-HD-9-0011]; the Liver Tissue Cell Distribution System (funded by NIH contract no. N01-DK-7-0004/HHSN267200700004C); and six samples were received from Xenotech, LLC (Lenexa, KS), to whom we are grateful for their generous gift of tissue samples. Thirty-four prenatal human liver samples (57–196 days, postconception, 15 female, 18 male, 1 unknown gender) were received from the Central Laboratory for Human Embryology (University of Washington, Seattle, WA, funded by the National Institute of Child and Human Development). All tissues were flash frozen and maintained at −80°C before isolation of DNA and RNA. All samples were from anonymous, deceased donors, and the use of these tissues was declared nonhuman subjects research by the University of Missouri—Kansas City Pediatric Health Sciences Review Board.
Methyl-Seq and Bisulfite Sequencing.
Genomic DNA was isolated from approximately 30 mg of pediatric livers using the AllPrep DNA/RNA mini kit (Qiagen, Valencia, CA). Determination of methylation status of cytosines within CpGs was performed with the SureSelectXT Human Methyl-Seq system (Agilent, Santa Clara, CA) for target enrichment on 16 pediatric liver samples, aged 20 days to 17 years. Libraries were sequenced on a HiSEq. 2000 (Illumina, San Diego, CA). Data were analyzed using the publicly available Bismark v0.7.4 (Krueger and Andrews, 2011) combined with Bowtie v0.12.8 (Langmead et al., 2009) to locate methylated CpG sites on both strands. Using inhouse software, gmcaller v0.10, methylated CpG sites were aggregated by individual followed by counting across chromosome positions. To each site, we attached its gene annotation and relative position to the translation start site.
Validation of methyl-seq results by bisulfite sequencing was performed on genomic DNA from all 48 pediatric liver samples. Sodium bisulfite conversion was performed on 1 μg of liver genomic DNA using the EpiTect Bisulfite kit according to the manufacturer’s recommendations (Qiagen). Bisulfite-treated DNA was used for polymerase chain reaction (PCR) amplification of the proximal promoter regions of CYP3A4 and CYP3A7 using primers designed with Bisearch online bisulfite primer design software (Table 1) (Tusnady et al., 2005). After nested PCR amplification, PCR products were treated with ExoSap to remove excess nucleotides and PCR primers. PCR products were sequenced via Sanger sequencing using the nested PCR primers. Sequencing chromatograms were analyzed using ab1 Peak Reporter (ThermoFisher, Waltham, MA) to generate numerical peak height data. Methylation at individual cytosines was calculated using Mquant as previously described (Leakey et al., 2008).
Primers used for targeted bisulfite sequencing
RNA-Seq.
Expression of CYP3A4 and CYP3A7 was determined with RNA-Seq on RNA derived from the same set of pediatric and prenatal liver samples on a HiSEq. 1500. RNA-Seq reads were mapped to hg19 reference genome using TopHat (Trapnell et al., 2009) integrated within Bowtie (v 0.12.8). Cufflinks v1.0.3 (Trapnell et al., 2010) was used to assemble liver transcriptomes from the aligned RNA-seq reads and quantifies their expressions, including those of CYP3A4 and CYP3A7.
Data Analysis.
Differential fractional methylation was analyzed and compared between age groups by using one-way analysis of variance (ANOVA) with Tukey’s post hoc test for pairwise comparisons between age groups. Correlation between mRNA expression and CpG methylation was examined using Spearman’s rank correlation coefficient. For all analyses, a P < 0.05 was considered significant.
Results
After target enrichment with SureSelect Human Methyl-Seq and next-generation sequencing, high-quality data were available for 39 cytosines in CpG dinucleotides across the CYP3A4 and CYP3A7 genes (of a total of 42 cytosines predicted for enrichment), representing 12% of all cytosines in CpG dinucleotides in the genomic sequences of CYP3A4 and CYP3A7. For CYP3A4, methylation levels were available for cytosines in the proximal promoter and introns 1 and 2, whereas for CYP3A7, methylation levels after target enrichment were available for the proximal promoter; introns 3, 4, and 5; and exons 5 and 13. Cytosine methylation from the promoter through intron 2 of CYP3A4 was highly variable (32%–96%) and did not exhibit a distinct pattern with respect to position along the gene sequence (Fig. 1A). The average percentage of methylation of cytosines through CYP3A7 (Fig. 1B) and the pseudogene CYP3AP1 was also highly variable across the entire region (38%–92%).

Methyl-seq determination of cytosine methylation in CYP3A4 and CYP3A7. Average fraction of methylated cytosines in pediatric liver as determined with methyl-seq analysis is shown with respect to position in the CYP3A4 (A) and CYP3A7 (B) genes. The Agilent SureSelect Human Methyl-Seq system enriches for a fraction of cytosines in the CYP3A4 and CYP3A7 genes. After target enrichment, next generation sequencing and data analysis were performed as described in Materials and Methods.
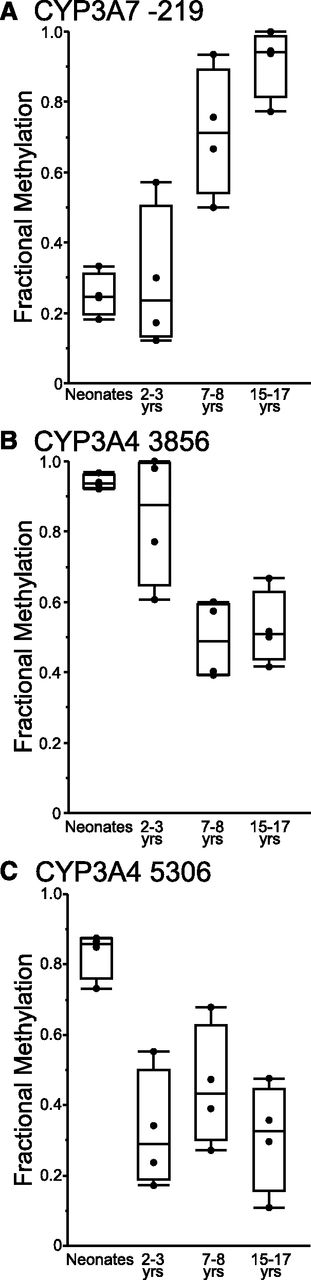
Changes in DNA methylation states of CpG sites within the CYP3A4 and CYP3A7 genomic sequences were investigated with respect to age. Samples used for methyl-seq analysis were assigned to one of four age groups: group 1 (infants 3–14 weeks of age), group 2 (children 2–3 years of age), group 3 (children 7–8 years of age), or group 4 (adolescents, 15–17 years of age). Average fraction methylation was compared between groups. Cytosines in the proximal promoter of CYP3A7 (at positions −182, −199, and −219 relative to the ATG start codon) exhibited statistically significant differences between the age groups (ANOVA, P < 0.005). For all three cytosines in the CYP3A7 proximal promoter, fractional methylation was lower in groups 1 and 2 than in groups 3 and 4. In contrast, fractional methylation of cytosine at −1042 of the CYP3A7 proximal promoter did not vary with respect to group. In addition, fractional methylation of a cytosine at position 14056 (relative to the ATG) in intron 3 of CYP3A7 was higher in group 1 relative to older children in groups 3 and 4 (P = 0.03). After Bonferroni correction for multiple hypothesis testing, however, the level of fractional methylation was significantly different by age group for CYP3A7 −219 only [(Fig. 2A) corrected P = 0.003]. No differences in fractional methylation between groups were observed for additional cytosines across the rest of the CYP3A7 and CYP3AP1 genes enriched by the SureSelect system.
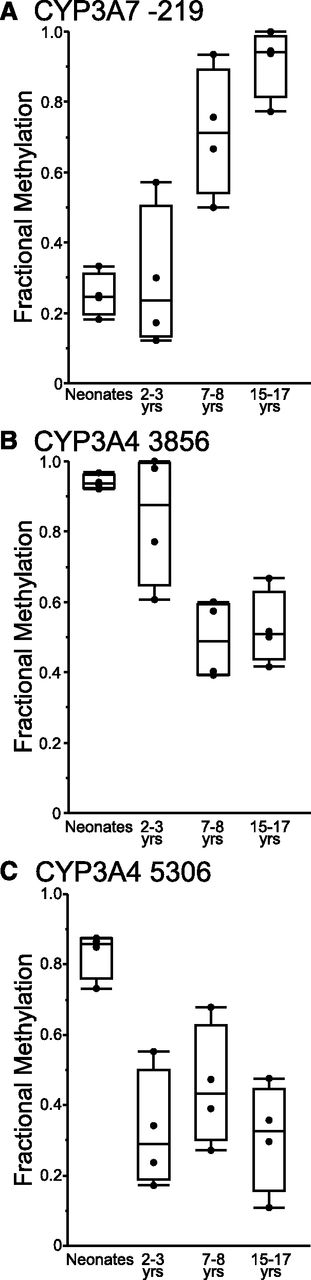
Results of methyl-seq analysis of the CYP3A4 and CYP3A7 genes. Cytosines that exhibit statistically significant changes in methylation levels of cytosines in the CYP3A7 proximal promoter (A) and in intron 1 (B) and intron 2 (C) of the CYP3A4 gene are shown.
No differences were observed in the fractional methylation of cytosines up to position −1569 of the proximal promoter of CYP3A4 with respect to group; however, differences in fractional methylation of cytosines in introns 1 and 2 of CYP3A4 (at positions 3856, 4951, 5059, and 5306) were observed between age groups (ANOVA, P < 0.05). Fractional methylation of cytosines within the introns 1 and 2 of the CYP3A4 gene decreased overall with postnatal age. After Bonferroni correction for multiple hypothesis testing, CYP3A4 −3856 (corrected P = 0.01) and CYP3A4 −5059 (corrected P = 0.04) exhibited statistically significant decreases in methylation with respect to postnatal age group (Fig. 2, B and C).
Multiple CpG sites in the CYP3A4 and CYP3A7 genes exhibited changes in fractional methylation that could be considered consistent with the expression patterns of the two enzymes in neonatal and pediatric human liver. To determine whether methylation levels were associated with expression of CYP3A4 or CYP3A7, fractional methylation was compared with full-length canonical mRNA transcript abundance (FPKM) determined by RNA-seq experiments performed on RNA isolated from the same human liver samples. Table 2 shows the Spearman correlation coefficients for statistically significant associations between CYP3A4 and CYP3A7 transcript abundance and methylation marks in the CYP3A4/7 genes. CYP3A7 mRNA expression was statistically significantly associated with methylation of CYP3A7 −219 and CYP3A7 −182, which also varied with postnatal age. CYP3A7 expression was also associated with methylation at sites in the CYP3A4 gene. Finally, expression of CYP3A4 mRNA was significantly associated with methylation levels in the proximal promoter at position −1547, a CpG that was associated with CYP3A4 expression in a previous study of adult livers (Kacevska et al., 2012).
Methylation correlated with transcript abundance
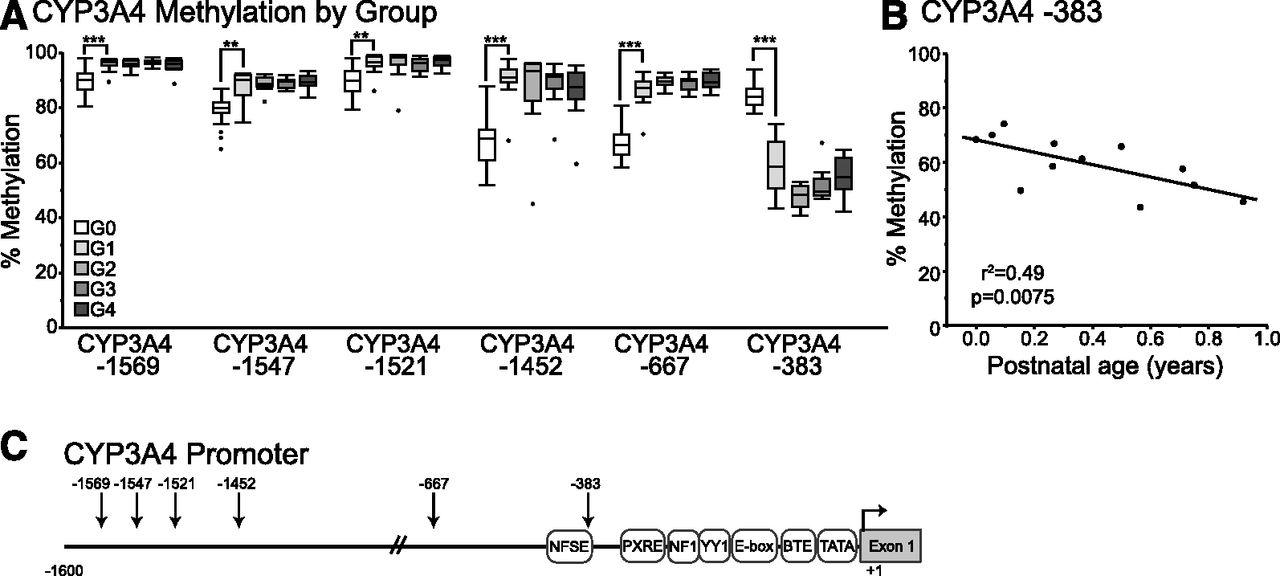
Results from methyl-seq data on a limited number of postnatal liver samples (n = 16) indicate that CpG dinucleotides within the CYP3A4 and CYP3A7 genes are differentially methylated during postnatal maturation in human livers. Furthermore, patterns of gene expression for CYP3A4 and CYP3A7 follow patterns of methylation, including modification of CpG dinucleotides in the proximal promoter regions of CYP3A7. Targeted bisulfite sequencing of the proximal promoters of CYP3A4 and CYP3A7 was performed on DNA isolated from 48 postnatal human liver samples (aged < 1 day to 17 years) to verify methyl-seq results. In addition, bisulfite sequencing was performed on 34 prenatal human liver samples to further characterize developmental changes in CYP3A4 and CYP3A7. For all cytosines in the CYP3A4 promoter covered by bisulfite sequencing, methylation levels in prenatal livers differed statistically from those observed in postnatal livers (Fig. 3A). Methylation of CYP3A4 −383 was higher in prenatal livers compared with those of children younger than 1 year of age (mean, 83% versus 59%, P < 0.0001), whereas lower levels of methylation were observed in prenatal livers compared with postnatal livers for CYP3A4 −667 (68% versus 87%, respectively, P = 0.0001), −1452 (69% versus 90%, P = 0.0005), −1521 (90% versus 96%, P = 0.003), −1547 (80% versus 88%, P = 0.003), and −1569 (89% versus 96%, P = 0.0005). Consistent with results from methyl-seq, bisulfite sequencing revealed that cytosine at CYP3A4 −383 is methylated at high levels in neonatal liver and exhibits statistically significant decreasing methylation levels with respect to age over the first year of life (Fig. 3B, r2 = 0.49, P = 0.0075). Interestingly, after 1 year of age, fractional methylation of CYP3A4 −383 increases in a linear manner with respect to age (r2 = 0.32, P = 0.0004).
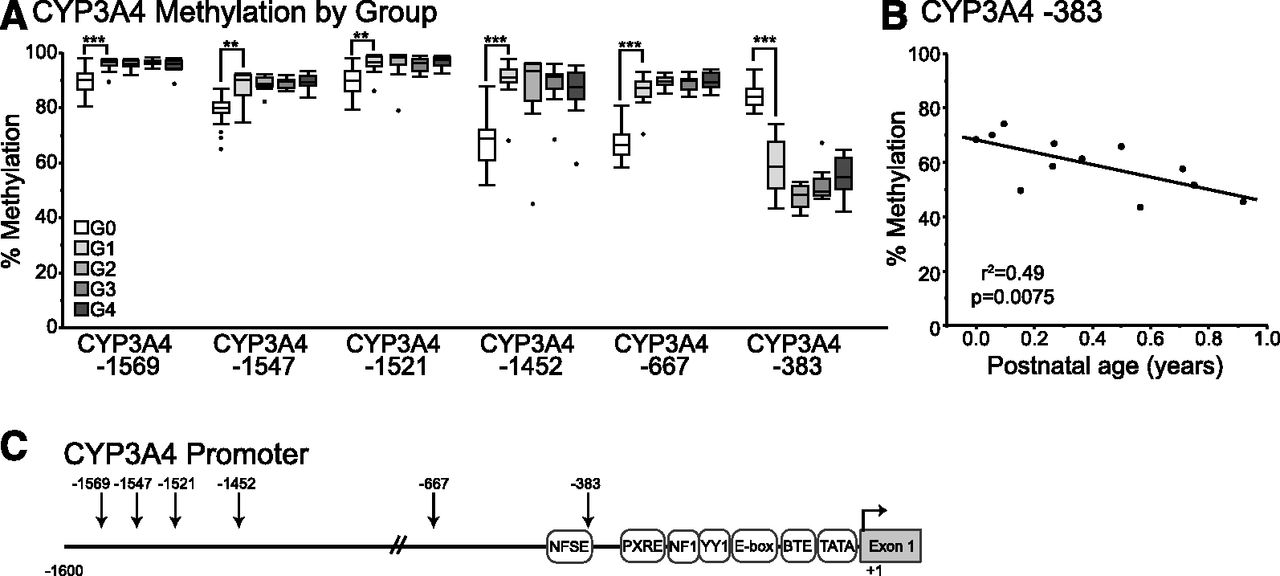
Results of targeted bisulfite sequencing of the CYP3A4 proximal promoter in prenatal and postnatal liver samples. Average percentage of methylation is shown versus age group where G0 = prenatal, G1 < 1 year of age, G2 = 1.8–6 years of age, G3 = 7–11 years of age, and G4 = 12–17 years of age. (A) Average percent methylation is shown by age group for each CpG in the proximal promoter of CYP3A4. (B) Methylation of cytosine at −383 of the CYP3A4 proximal promoter exhibits a decrease in methylation over the first year of life. Significant differences in prenatal versus age group G1 are shown as **P < 0.01, ***P < 0.001. (C) The CYP3A4 proximal promoter region is shown with the positions of known regulatory elements indicated. The positions of CpG dinucleotides examined with targeted bisulfite sequencing are indicated with arrows.
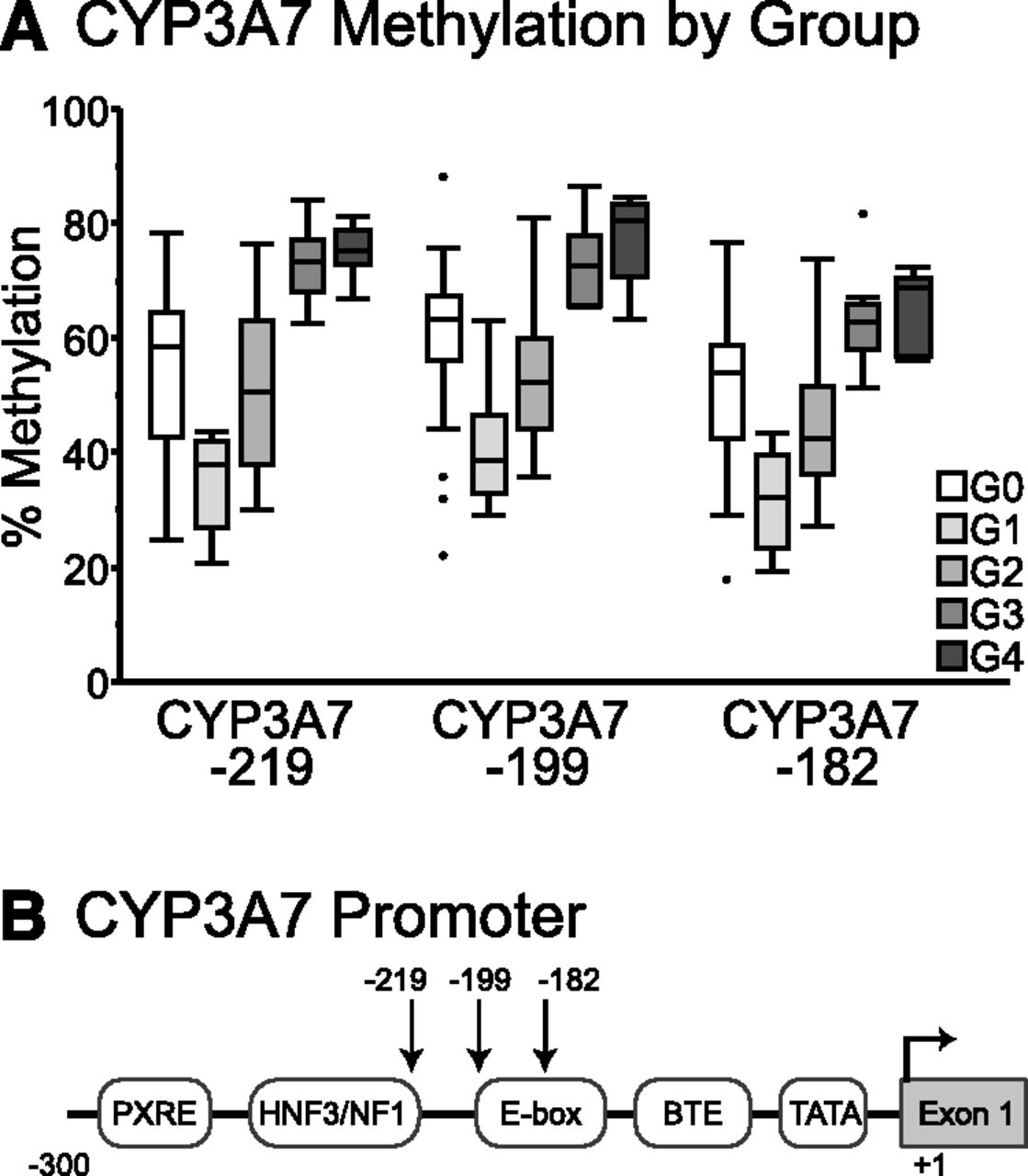
Bisulfite sequencing of the proximal promoter of CYP3A7 also confirmed results of methyl-seq analysis in postnatal human livers with statistically significant differences in methylation levels between age groups for all three cytosines at CYP3A7 −182, −199, and −219 (Fig. 4A). Interestingly, DNA from prenatal liver revealed that methylation levels of the CYP3A7 proximal promoter were higher in prenatal samples than those observed in samples from infants, seemingly in contrast to higher levels of CYP3A7 gene expression in prenatal versus postnatal liver. Furthermore, a decrease in methylation levels of cytosines in the CYP3A7 proximal promoter was observed over the first year of life, after which methylation levels increased.

Results of targeted bisulfite sequencing of the CYP3A7 proximal promoter in prenatal and postnatal liver samples. (A) Average percentage of methylation is shown by age group where G0 = prenatal, G1 < 1 year of age, G2 = 1.8–6 years of age, G3 = 7–11 years of age, and G4 = 12–17 years of age for three cytosines in the CYP3A7 proximal promoter. (B) The CYP3A7 proximal promoter region is shown with the positions of known regulatory elements indicated. The positions of CpG dinucleotides examined with targeted bisulfite sequencing are indicated with arrows.
Methylation of cytosines in the promoter and 5′ sequences of genes is associated with the repression of gene expression. Methylation levels of the proximal promoters of CYP3A4 and CYP3A7 were compared with expression levels determined via RNA-seq. In the full set of prenatal and postnatal liver samples, the expression of CYP3A7 was inversely correlated with methylation of CYP3A7 −219, CYP3A7 −199, and CYP3A7 −182 (Table 3; Supplemental Fig. 1). CYP3A4 expression was inversely correlated with CYP3A4 −383 (Spearman’s ρ = −0.48, P = 0.0001) and positively correlated with CYP3A4 −667, CYP3A4 −1452, CYP3A4 −1521, CYP3A4 −1547, CYP3A4 −1569 (Table 3; Supplemental Fig. 2). When stratified by prenatal and postnatal age groups, CYP3A7 mRNA expression was inversely correlated with methylation of the proximal promoter in postnatal livers but not in prenatal livers. In postnatal livers, CYP3A4 mRNA expression was positively correlated with methylation of CYP3A4 −1569 (Spearman’s ρ = 0.3, P = 0.04) but not with any other cytosine in the proximal promoter. CYP3A4 expression was not associated with methylation levels of cytosines in the proximal promoter in prenatal liver.
Spearman’s correlation coefficients between expression and methylation
Discussion
Here we report the differential methylation of cytosines in the human CYP3A4 and CYP3A7 genes between prenatal and postnatal human liver. Previous studies have shown that high levels of CpG methylation in the 5′ regulatory regions and 5′ exons and introns are associated with repression of gene expression. Patterns of cytosine methylation of the CYP3A4 and CYP3A7 proximal promoter regions are consistent with established developmental changes in gene expression. Interindividual variability in CYP3A4 mRNA expression in postnatal liver was modestly associated with methylation at CYP3A4 −1569.
The developmental transition between CYP3A7 and CYP3A4 in human liver has been well-documented; however, the mechanisms that drive developmental expression of the two enzymes remain unknown. There is a high degree of sequence identity (> 90%) between the genes encoding human CYP3A4 and CYP3A7 that extends 9 kb upstream of the two genes, including the proximal promoter regions and the distal xenobiotic responsive enhancer module regulated by the nuclear receptors PXR and constitutive androstane receptor (CAR). CYP3A7 has been shown to be expressed at significant levels in approximately 10% of adult tissues, and expression is predominantly associated with the CYP3A7*1C polymorphism and, to a lesser extent, the CYP3A7*1B polymorphism (Burk et al., 2002; Sim et al., 2005). The CYP3A7*1C allele is characterized by a region between −291 and −232 bp of the proximal promoter that has been replaced with the corresponding sequences of the CYP3A4 gene promoter. The molecular mechanism by which CYP3A7*1C leads to high levels of expression in adult tissue is attributed to increased activation of the CYP3A7*1C promoter by the nuclear receptors PXR and CAR (Burk et al., 2002). The association of proximal promoter polymorphisms of CYP3A7 with its expression in adult tissues implicates this region as a potentially important determinant of developmental regulation.
Results from transgenic mouse models have been used to understand the regulation of CYP3A4 and CYP3A7. Two separate transgenic mouse models have been developed that carry reporter genes (lacZ and luciferase) under the control of approximately 13 kb of the human CYP3A4 5′ regulatory region (Robertson et al., 2003; Zhang et al., 2003). In both studies, reporter gene expression was strongly induced by PXR ligands. Robertson et al. (2003) also reported that mice carrying only 3.2 kb of the CYP3A4 5′ regulatory region had essentially no expression of the lacZ reporter in the liver and small intestine in the absence or presence of inducers and concluded that elements upstream of 3.2 kb were necessary to direct tissue-specific expression. In transgenic mice carrying the entire human CYP3A4 gene, including > 8 kb of the 5′ promoter, basal expression of CYP3A4 was reported in mouse small intestines but not in mouse livers, further suggesting that distal regulatory elements are necessary for high levels of hepatic expression of CYP3A4 (Granvil et al., 2003). Additional transgenic mouse lines carrying the complete genomic sequences of human CYP3A4 and CYP3A7, including 35 kb upstream of CYP3A4, 9.5 kb downstream of CYP3A7, and the entire intergenic region, have been generated (Cheung et al., 2006; Ma et al., 2008; Hasegawa et al., 2011; Pang et al., 2012). These mice exhibit expression of CYP3A4 and CYP3A7 in liver that is consistent with developmental patterns of expression in human liver (Pang et al., 2012). These data suggest that genomic elements that determine basal and developmental changes in gene expression of these two enzymes are encompassed within this region.
Despite the high sequence identity, CpG dinucleotides in the proximal promoters of CYP3A4 and CYP3A7 that are shown here to be differentially methylated during prenatal/postnatal development are not conserved between the two genes; however, comparison between human and chimp CYP3A promoter sequences reveal that these CpG dinucleotides are evolutionarily conserved between human and chimp orthologs. Furthermore, differentially methylated CpG dinucleotides of the CYP3A7 proximal promoter are found within known and predicted regulatory elements of the promoter. Functional studies comparing transcription factor binding and function of the CYP3A4 and CYP3A7 proximal promoters show a complex interplay between transcription factors leading to differential transcription initiation in transient transfected HepG2 cells (Saito et al., 2001). Liver-enriched transcription factors NF1, HNF3β, and USF1 are capable of binding to the DNA sequences of the CYP3A7 proximal promoter (Fig. 4B) that are differentially methylated during development. Whereas USF1 and NF1 are also capable of binding to the corresponding sequences of the CYP3A4 proximal promoter (Fig. 3C), HNF3β does not bind, whereas YY1 is capable of binding adjacent sequences of the CYP3A4 promoter. In addition, we have shown that members of the NF1 family bind to the region of the CYP3A7 proximal promoter that is differentially methylated during development as well as to the similar sequences from the CYP3A4 promoter (Riffel et al., 2009); however, members of the NF1 transcription factor family differentially affect the activity of the CYP3A4 and CYP3A7 proximal promoters in reporter assays in transiently transfected HepG2 cells whereby the transcriptional activity of the CYP3A4 promoter is increased by members of the NF1 family and the transcriptional activity of the CYP3A7 proximal promoter is decreased (Riffel et al., 2009).
In contrast to methylated cytosines in the CYP3A7 proximal promoter, it is less clear how methylated cytosines in the CYP3A4 promoter may alter gene expression (Fig. 3C). The cytosine at −383 is found within the nifedipine response element (NFSE). Nuclear proteins are capable of binding to the NFSE of CYP3A4; however, the identity and the contribution these proteins make to the regulation of CYP3A4 are not known (Hashimoto et al., 1993; Westlind et al., 1999). The CYP3A4*1B polymorphism is an A→G variation just upstream of CYP3A4 −383 that also affects the NFSE sequence, but the functional consequences of this variant on CYP3A4 expression are controversial (Rebbeck et al., 1998; Westlind et al., 1999; Rodriguez-Antona et al., 2005; Klein et al., 2012). The remaining differentially methylated cytosines of the CYP3A4 promoter occur in sequences that have not yet been associated with transcription factor binding. Given the association of methylation at CYP3A4 −1569 with the expression of CYP3A4 mRNA, however, additional studies investigating the role of these regions and the role of DNA methylation on transcription factor binding and promoter activity should be considered.
The apparently high levels of cytosine methylation in the CYP3A7 proximal promoter in prenatal human liver would seem to be in conflict with high levels of CYP3A7 expression. This observation may be due to the nature of prenatal liver, which is the primary site of hematopoiesis and, as such, is dominated by the presence of hematopoietic stem cells (HSC) rather than parenchymal hepatocytes as found in the adult liver. Previous studies have shown that CYP3A7 is not expressed by HSCs of fetal liver (Shao et al., 2007); therefore, the predominance of HSC (∼50% of liver mass at midgestation) (Zaret, 2000) in fetal liver where CYP3A7 is not expressed may mask actual CpG methylation levels in parenchymal hepatocytes, resulting in the high levels of cytosine methylation in the CYP3A7 promoter that are reported here. Alternatively, bisulfite conversion does not distinguish between 5-methylcytosines and 5-hydroxymethylcytosines and high levels of CpG methylation observed here might be due to 5-hydroxymethylcytisines in the CYP3A7 promoter. Whereas 5-methylcytosine in the 5′ regulatory region of a gene is associated with gene silencing, 5-hydroxymethylcytosines are associated with gene expression. Furthermore, 5-hydroxymethylcytosine is believed to be an intermediate of DNA demethylation formed by members of the 10–11 translocation family that oxidize 5-methylcytosine to 5-hydroxymethylcytosine.
In conclusion, we have shown developmental changes in human pediatric livers in the methylation of cytosines within the genes encoding human CYP3A4 and CYP3A7 using next-generation sequencing after target enrichment. Changes in methylation of the proximal promoters of CYP3A4 and CYP3A7 were confirmed with targeted bisulfite sequencing and were shown to be associated with developmental changes in expression of the enzymes in human prenatal and pediatric livers; however, the functional consequences of differential methylation on transcription factor binding and transcription initiation from the CYP3A4 and CYP3A7 promoters remain undefined. Therefore, further studies required to determine the functional mechanism by which alterations in methylation lead to changes in gene expression for CYP3A4 and CYP3A7 are ongoing.
Acknowledgments
The authors thank Darrell Dinwiddie, Mohammad Rezaiekhaligh, and Emily Fiore for their technical expertise.
Authorship Contributions
Participated in research design: Vyhlidal, Bi, Ye, Leeder.
Conducted experiments: Vyhlidal, Ye.
Performed data analysis: Vyhlidal, Bi.
Wrote or contributed to writing of the manuscript: Vyhlidal, Bi, Ye, Leeder.
Footnotes
- Received December 1, 2015.
- Accepted January 14, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ANOVA
- analysis of variance
- bp
- base pair
- CAR
- constitutive androstane receptor
- CpG
- cytosine-phospho-guanine
- HSC
- hematopoietic stem cells
- kb
- kilobase
- PCR
- polymerase chain reaction
- PXR
- pregnane X receptor
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}