


Abstract
11-Ethyl-5,11-dihydro-5-methyl-8-[2-[(1-oxido-4-quinolinyl)oxy] ethyl]-6H-dipyrido[3,2-b:2′,3′-e][1,4]diazepin-6-one (BILR 355) is an inhibitor of the human immunodeficiency virus-1. BILR 355 exhibited a nonlinear pharmacokinetic profile and low exposure after oral administration to humans. This article describes the in vitro metabolism of BILR 355, which is correlated with the in vivo nonlinearity findings. Our in vitro studies had demonstrated that BILR 355 was extensively metabolized by cytochrome P450 3A. Thus, BILR 355 was concomitantly administered with ritonavir (RTV) in an attempt to boost systemic exposure, which did occur in humans. In addition, the expectation was that the overall metabolism of BILR 355 would be decreased with concomitant administration of RTV. Subsequent metabolite profiling was performed using human plasma samples obtained from clinical phase Ib studies with concomitant administration of BILR 355 and RTV. A total of 18 metabolites was observed. Their structures were proposed on the basis of high-performance liquid chromatography-tandem mass spectrometry technologies, and 10 metabolites were confirmed by comparison with synthetic standards. We were surprised to find that a disproportionate human metabolite, BILR 516, was uncovered during this metabolite profiling study and pharmacokinetic analysis of BILR 516 showed that it had a longer half-life and higher exposure than the parent compound at steady state. Of interest, BILR 516 was not detected in human plasma when BILR 355 was administered alone. Therefore, whereas RTV boosted the exposure of BILR 355, it resulted in a significant metabolic switching of BILR 355. Overall, this article demonstrates an unusual example of metabolic switching and raises concern about the consequence of metabolic switching during drug development.
Introduction
Human immunodeficiency virus (HIV)-1 infection remains a major worldwide health issue. The current antiretroviral therapy has dramatically reduced morbidity and mortality among HIV-infected patients and significantly modified the course of HIV disease into a manageable chronic disease with longer survival and improved quality of life for patients (Hogg et al., 1998; Palella et al., 1998; Moreno et al., 2010). However, resistance has emerged to almost all of these antiretroviral agents, sometimes rendering them ineffective (Hammer et al., 2008). First-generation non-nucleoside reverse transcriptase inhibitors (NNRTIs) are particularly vulnerable, because resistance may emerge with a single resistance codon alteration and the emergence of drug-resistant HIV-1 variants is fairly rapid (De Clercq, 1998). In addition, extensive cross-resistance among NNRTIs was observed, resulting in a loss of the efficacy of subsequent NNRTI treatment (de Béthune, 2010). Therefore, development of additional NNRTIs to effectively treat patients with NNRTI-resistant HIV is essential (De Clercq, 2004).
11-Ethyl-5,11-dihydro-5-methyl-8-[2-[(1-oxido-4-quinolinyl)oxy] ethyl]-6H-dipyrido[3,2-b:2′,3′-e][1,4]diazepin-6-one (BILR 355) is a second-generation NNRTI (Bonneau et al., 2005; Boone, 2006). The in vitro results showed that the EC50 of BILR 355 was 0.26 ng/ml (0.59 nM) against wild-type HIV-1 and ranged from 1.5 to 13 ng/ml (3.4–29 nM) against clinically common single and double NNRTI mutations (Bonneau et al., 2005). A clinical study revealed that BILR 355 was rapidly absorbed after a single oral dose in healthy volunteers with the mean time to maximum drug concentration (Tmax) of 0.5 to 1.5 h. However, thereafter the concentrations of BILR 355 rapidly declined, resulting in a short terminal half-life (t1/2) of approximately 2 to 4 h (Huang et al., 2008). Our in vitro data demonstrated significant metabolism of BILR 355 by cytochrome P450 (P450) 3A. Thus, in an attempt to overcome the resulting low exposure in humans, BILR 355 was administered with ritonavir (RTV), a potent CYP3A inhibitor previously used to boost plasma concentrations and to increase the t1/2 of HIV protease inhibitors (Hull and Montaner, 2011). With concomitant administration of RTV, the t1/2 of BILR 355 increased to approximately 10 to 16 h and the maximum plasma concentration (Cmax) increased 2- to 5-fold with the area under the plasma concentration-time curve from time 0 to infinity (AUC0–∞) increasing 15- to 30- fold (Huang et al., 2008), a profile more consistent with once daily dosing.
However, this combination created an unexpected problem, a disproportionate human metabolite (BILR 516), as a result of significant metabolic switching of BILR 355. The current article describes the in vitro metabolism of BILR 355 by CYP3A and identification of the metabolites resulting from metabolic switching due to inhibition of CYP3A by concomitantly administered RTV.
Materials and Methods
Chemicals, Reagents, and Other Materials.
BILR 355, BILR 402, BILR 516, INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, BI 212580, and BI 212578 were synthesized at Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT), and their structures are shown in the supplemental data. BILR 483 (D3-BILR 355 with three deuterium atoms on the methyl group) and nevirapine were also synthesized at Boehringer Ingelheim Pharmaceuticals, Inc. NADPH was purchased from Sigma-Aldrich (St. Louis, MO). All other reagents and solvents were of analytical grade or higher purity and were obtained from commercial suppliers. Human liver microsomes (HLMs) from individual donors and pooled HLMs from 15 donors were obtained from Celsis In Vitro Technologies (Baltimore, MD). All recombinant P450s (produced in baculovirus-infected insect cells) and control insect cell microsomes were obtained from BD Biosciences (Woburn, MA). The microsomes and recombinant P450s were stored at −80°C until used.
Km and Vmax Determination of BILR 355 Metabolism.
BILR 355 was incubated with HLMs from four individual donors to assess apparent Km and Vmax values. Incubations were performed in 0.1 M potassium phosphate buffer (containing 5 mM MgCl2, pH 7.4). Microsomes were preincubated with BILR 355 for 5 min at 37°C. The reaction was initiated by the addition of NADPH at a final concentration of 2 mM. The final concentrations of microsomes were 0.5, 1, or 2 mg/ml. The final incubation volume was 0.9 ml, and the final organic solvent concentration in each reaction did not exceed 1%. Aliquots (100 μl) were transferred from the incubation mixtures at various time intervals (0, 0.25, 1, 2, 3, 5, 10, 15, and 30 min) and quenched with an equal volume of acetonitrile. Then, 100 μl of BILR 483 in acetonitrile at a concentration of 0.5 μM was added as the internal standard to each well. All samples were centrifuged at 1600g for 10 min, and the supernatants were analyzed by LC-MS/MS. The experiment was performed in duplicate for each lot of HLMs.
Metabolism of BILR 355 by Recombinant P450 Isoforms.
BILR 355 (1 or 10 μM) was incubated with recombinant P450 isoforms (rCYP1A2, rCYP2B6, rCYP2C9, rCYP2C19, rCYP2D6, and rCYP3A4) in 0.1 M potassium phosphate buffer with 5 mM MgCl2 at pH 7.4. The final recombinant P450 concentrations were 50 pmol/ml, and the total incubation volume was 800 μl. The reactions were preincubated at 37°C for 5 min before the addition of NADPH at a final concentration of 2 mM. Control samples were prepared identically, except that nontransfected insect cell microsomal protein was substituted for individual recombinant P450 isoforms. The depletion of BILR 355 was monitored over a full time course of 30 min. Aliquots of incubation mixtures were transferred at 0.5, 2, 5, 10, 15, 20, and 30 min and quenched with acetonitrile containing BILR 483 as internal standard. The plates were centrifuged and the supernatant was analyzed by LC-MS/MS. The experiment was performed in duplicate.
Inhibition of BILR 355 Metabolism in HLMs by Isoform-Selective Inhibitors.
BILR 355 (1 μM) was incubated with pooled HLMs in the presence and absence of isoform-selective chemical inhibitors. The chemical inhibitors were furafylline (30 μM) for CYP1A2, sulfaphenazole (5 μM) for CYP2C9, tranylcypromine (100 μM) for CYP2C19, quinidine (5 μM) for CYP2D6, and ketoconazole (1 μM and 3 μM) for CYP3A4 (Bourrié et al., 1996; Chauret et al., 1997). The incubation conditions and sample analysis were as described above for HLMs. The experiment was performed in triplicate.
Metabolite Profiling of BILR 355 in Clinical Samples.
Human plasma samples were obtained from six healthy male volunteers administered 150 mg of BILR 355 with 100 mg of RTV b.i.d. (clinic trial number 1188.2, cohort 12, conducted at the Buffalo Clinical Research Center in Buffalo, NY) (Huang et al., 2009). Steady-state levels of BILR 355 were achieved by day 7, and only one dose of BILR 355 and RTV was given on day 7 to obtain a full PK profile. Intensive PK samples were taken on day 1 at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 9, and 12 h and on day 7 at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 9, 12, 16, 24, 48, and 72 h after drug administration. Plasma samples from different subjects were pooled at Tmax (1.5 h) and two later time points (6 and 24 h) on day 7. The 0-h samples at day 1 were also pooled and used as control samples for metabolite identification. The pooled plasma samples were vortexed thoroughly, and 300-μl aliquots were transferred to Eppendorf tubes. Nine hundred microliters of methanol was added to the plasma aliquot. The mixture was vortexed thoroughly and centrifuged at 17,000g for 5 min. The supernatant was then dried down under nitrogen at room temperature. The residue was reconstituted in 200 μl of water-methanol (50:50; v/v). The reconstituted sample was centrifuged at 17,000g for 5 min, and the supernatant was used for metabolite identification by LC-MS/MS. After the metabolites were identified, nine metabolites with authentic standards, including INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, BILR 516, BI 212578, and BI 212580, were quantitated by LC-MS/MS using the plasma samples at selected time points. Nevirapine was used as the internal standard at a final concentration of 0.1 μM for metabolite quantitation.
Equipment and Chromatographic Conditions for Quantitation and Metabolite Identification of BILR 355.
For quantitation of BILR 355 in in vitro incubations, an LC-MS/MS system consisting of a Gilson 215 liquid handler autosampler (Gilson, Inc., Middleton, WI), two series 200 Micro pumps (PerkinElmer Life and Analytical Sciences, Waltham, MA), and a Applied Biosystems MDS Sciex API365 triple quadrupole mass spectrometer (Applied Biosystems/Sciex, Thornhill, ON, Canada) was used. The column used was a Symmetry C18 (2 × 50 mm, 3.5-μm particle size; Waters, Milford, MA). Mobile phase compositions were as follows: mobile phase A, water-acetonitrile-acetic acid (95:5:0.05; v/v/v); and mobile phase B, water-acetonitrile-acetic acid (5:95:0.05 v/v/v). BILR 355 and the isotope-labeled internal standard, BILR 483, eluted at 1.7 min with a gradient of mobile phase B (maintain at 15% B for 0.5 min, increase from 15% B to 85% B in 3 min, and then keep at 85% B for 0.5 min) at a flow rate of 0.25 ml/min. The mass spectrometer was optimized for BILR 355 with an IonSpray voltage of 5.2 kV, an ion source temperature of 350°C, an orifice plate voltage of 35 V, a ring voltage of 300 V, and nebulizer gas of 13 l/min. The multiple reaction monitoring transitions requested for BILR 355 and the internal standard BILR 483 were m/z 442 → 281 and m/z 445 → 284, respectively.
Different LC-MS/MS instruments were used for identification and quantitation of metabolites of BILR 355. The instruments consisted of an SIL-5000 autosampler and two LC-10AD vp pumps (Shimadzu Scientific Instruments, Norwell, MA) connected with a Applied Biosystems MDS Sciex 4000 QTrap mass spectrometer (Applied Biosystems/Sciex). An Atlantis dC18 column (3.9 × 150 mm, 3-μm particle size; Waters) was used. Mobile phases were similar to those described previously except that the concentration of acetic acid was 0.1%. For metabolite identification, the gradients were the following: mobile phase B 0 to 1% over 10 min, 1 to 3% over 1 min, 3 to 12% over 19 min, 12 to 20% over 20 min, 20 to 29% over 25 min, 29 to 70% over 8 min, and 70 to 100% over 2 min at a flow rate of 0.7 ml/min. For metabolite quantitation of INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, and BILR 516, a 20-min gradient with mobile phase B increasing from 15 to 55% was used at a flow rate of 0.7 ml/min. The multiple reaction monitoring transitions requested and the retention times for metabolites are as follows: INLA 1678 (m/z 412 → 267; 7.3 min), BI 211136 (m/z 299 → 271; 7.6 min), BIRK 122 (m/z 299 → 271; 9.0 min), EXLS 1451 (m/z 414 → 253; 9.7 min), INRF 105 (m/z 313 → 285; 9.9 min), BILR 564 (m/z 428 → 267; 10.9 min), and BILR 516 (m/z 442 → 281; 16.1 min). The internal standard nevirapine was monitored at m/z 267 → 226 (8.0 min). For metabolite quantitation of BI 212580 and BI 212578, a 5-min gradient was used (mobile phase B increased from 45 to 55% in 5 min at a flow rate of 0.7 ml/min). The multiple reaction monitoring transitions for BI 212580, BI 212578, and the internal standard nevirapine were m/z 458 → 253, m/z 428 → 267, and m/z 267 → 226, respectively, and their retention times were 2.5, 3.5, and 2.8 min, respectively. The mass spectrometer parameters for the above analyses included an IonSpray voltage at 4.5 kV, a declustering potential at 50 V, an ion source temperature at 550°C, and collision energy at 30 V.
Data Analysis for Kinetic Parameters and Clearance Determination.
Apparent kinetic parameters for BILR 355 were determined using substrate depletion. The initial rate of BILR 355 depletion in HLMs was estimated using a linear regression of compound remaining at several initial time points demonstrating an acceptable linearity with r2 > 0.9. The apparent kinetic parameters for the depletion of BILR 355 by each HLM lot were determined by nonlinear regression analysis based on the Michaelis-Menten equation using the statistical program GraFit (version 5; Erithacus Software Ltd., Horley, UK). The Km and Vmax values obtained were used to calculate the intrinsic clearance (CLint, in vitro) according to CLint, in vitro = Vmax/Km, where Vmax is the normalized maximum reaction velocity in picomoles per minute per milligram of human liver microsomal protein and Km is the apparent Michaelis-Menten constant (Obach et al., 1997). The following constants were used to estimate CLint, in vivo from CLint, in vitro: a value of 45 mg of microsomal protein/g liver (Houston, 1994), 1800g of human liver weight, and average 70 kg of human body weight (Davies and Morris, 1993). To calculate the hepatic clearance, the well stirred model was applied, and hepatic clearance (CLh) (milliliters per minute per kilogram) was calculated using the following equation (Pang and Rowland, 1977):
 where Qh is the hepatic blood flow in humans (20.7 ml · min−1 · kg−1) (Davies and Morris, 1993) and fu is the fraction of unbound drug (assume fu = 1).
where Qh is the hepatic blood flow in humans (20.7 ml · min−1 · kg−1) (Davies and Morris, 1993) and fu is the fraction of unbound drug (assume fu = 1).
Results
Apparent Michaelis-Menten Kinetic Parameters of BILR 355 Metabolism in HLMs.
Four lots of precharacterized HLMs were selected on the basis of their testosterone 6β-hydroxylase activities representing a range from low to high CYP3A4 activities from available individual lots of human liver microsomes. Protein linearity was evaluated. Rates of metabolism were linear with concentrations of HLMs over the range of 0.5 to 2 mg/ml microsomal protein. Therefore, the amount of microsomal protein in the Km and Vmax assay was adjusted within the range of 0.5 to 2 mg/ml, so that the initial rate of the metabolism could be practically measured for each lot of HLMs. The initial rate of BILR 355 depletion was measured at various substrate concentrations (0.25, 0.5, 1, 2, 4, and 8 μM). The apparent Km and Vmax values for each HLM lot were calculated using nonlinear regression analysis based on the Michaelis-Menten equation. The mean Km and Vmax values are 0.79 μM and 111 pmol · min−1 · mg−1, respectively, as summarized in Table 1. Intrinsic clearance and hepatic clearance values calculated from each lot of HLMs are also listed in Table 1.
Michaelis-Menten kinetic parameters for BILR 355 metabolism as measured by compound depletion in human liver microsomes (n = 2)
P450 Reaction Phenotyping.
The metabolism of BILR 355 by various recombinant P450 isoforms (CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) was evaluated by measuring the time-dependent compound depletion in the presence of NADPH. BILR 355 was significantly metabolized only by recombinant human CYP3A4. The initial rate of BILR 355 depletion was calculated from a linear regression of compound remaining at several initial time points demonstrating an acceptable linearity with r2 > 0.9. The average initial rate of BILR 355 depletion by rCYP3A4 was 2.50 ± 0.21 and 2.59 ± 0.02 nmol · min−1 · nmol−1 P450 at the initial substrate concentrations of 1 and 10 μM, respectively. Isoform-selective inhibitors of the five major drug-metabolizing P450 isoforms were used to investigate their inhibitory effects on BILR 355 metabolism by HLMs. In the presence of a selective inhibitor of CYP3A4, ketoconazole at 1 and 3 μM, the metabolism of BILR 355 was inhibited by approximately 70 and 100%, respectively. The other chemical inhibitors did not cause any significant inhibitory effects. Furthermore, as shown in Table 1, Vmax of BILR 355 metabolism with HLMs ranged from 35 to 230 pmol · min−1 · mg−1 microsomal protein. The Vmax values correlated well with the testosterone 6β-hydroxylase activities (Table 1) of each HLM lot reported from the vendor (r2 = 0.96). There were no correlations with other reported P450-selective probe activities, including phenacetin O-deethylation activity (CYP1A2), coumarin 7-hydroxylase activity (CYP2A6), mephenytoin 4-hydroxylase activity (CYP2C19), and dextromethorphan O-demethylation activity (CYP2D6) (data not shown).
Identification of Metabolites of BILR 355 in Clinical Samples.
A total of 18 metabolites were detected, and a representative chromatogram of the samples is shown in Fig. 1, after molecular ion extraction of the identified metabolites from a full scan chromatogram. Only eight metabolites are shown in Fig. 1. The peak intensities of the other metabolites are too low, and thus their peaks are not visible in the ion extraction chromatogram. The metabolites are labeled M1 through M18 according to their respective order of elution from the HPLC column (Table 2). The possible structures of the metabolites are proposed on the basis of their molecular ions and product ion spectra. Because of space limitation, only the product ion spectra of the parent and the three metabolites of interest are shown in Fig. 2. The fragmentation of the parent and the rationale to elucidate the structures of the three metabolites are described below.

Representative LC-MS/MS chromatogram with extracted ions of the identified metabolites.
List of retention times, molecular ions, and characteristic product ions of BILR 355 and its metabolites

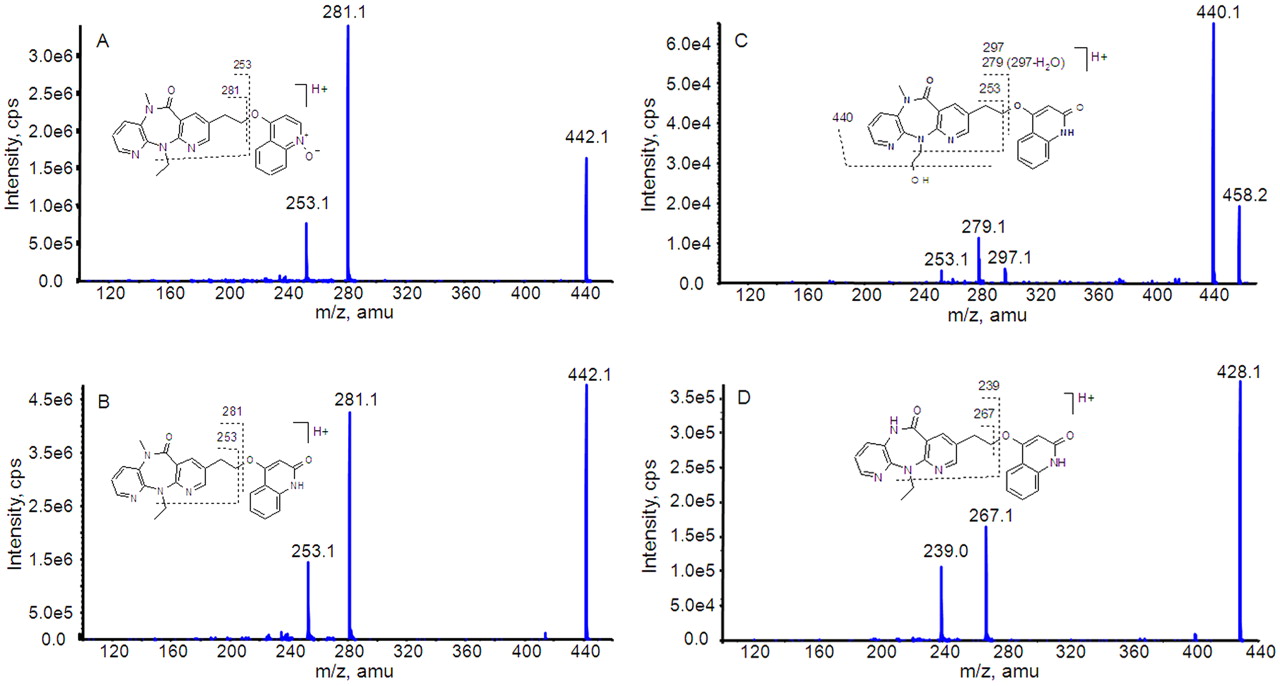
Product ion spectra of BILR 355 (A) and three metabolites, M18 (B), M14 (C), and M17 (D).
BILR 355.
BILR 355 eluted from the HPLC column with a retention time of 70.0 min. The product ion MS/MS spectrum of BILR 355 (Fig. 2A) was used as a reference to compare with the metabolite spectra. The protonated molecular ion of BILR 355 had m/z of 442. The product ion spectrum contains base ions at m/z 281 and 253. The product ion at m/z 281 arises from the loss of the quinoline N-oxide from the protonated parent ion. This is followed by a subsequent loss of the ethyl group from the diazepinone ring, resulting in an ion at m/z 253.
M18.
The peak of the metabolite M18 eluted at approximately 79.9 min. This metabolite had a protonated molecular ion at m/z 442, which was the same as that of the parent compound. However, this metabolite eluted later than the parent compound, suggesting that it was an isomer of BILR 355. The product ion spectrum of M18 yielded the same product ions at 281 and 253 as the parent compound (Fig. 2B), which indicated that the molecular composition of the dipyridodiazepinone moiety and the quinoline moiety remained unchanged. It is known that N-oxides are susceptible to reduction; therefore, it is possible that the N-oxide is reduced and further oxidation could occur at a position other than the nitrogen on the quinoline moiety, resulting in the formation of an isomer of BILR 355. The structure of M18 was further confirmed as BILR 516 based on the comparison with an authentic standard. The metabolite and the authentic standard had the same retention time, molecular weight, and MS/MS pattern.
Similar to the relationship between BILR 355 and BILR 516, two other pairs of metabolites were identified, i.e., M7–M14 and M13–M17. Each pair of metabolites had the same protonated molecular ions and the same product ions, but different retention times. Therefore, it was proposed that M7 and M13, which eluted earlier, had the quinoline N-oxide structure similar to BILR 355, and M14 and M17, which eluted later, had the quinolone structure similar to BILR 516.
M14.
This metabolite had a protonated molecular ion at m/z 458, which is 16 amu higher than that of BILR 516, suggesting an addition of oxygen. Its product ion spectrum showed abundant product ions at m/z 253, 279, 297, and 440 (Fig. 2C). The product ion at m/z 297 indicated possible oxidation on the dipyridodiazepinone moiety. Two abundant product ions at m/z 440 and m/z 279 could originate from the loss of H2O from the molecular ion and the product ion at m/z 297, respectively. Similar to BILR 516, a product ion at m/z 253 was detected for M14, which suggested the dipyridodiazepinone ring was unchanged. Thus, the likely oxidative position would be the ethyl group connected to the diazepinone ring. In addition, oxidation at the terminal carbon is relatively more stable compared with the oxidation at the α-position. Therefore, M14 was tentatively identified as a hydroxylated BILR 516 with oxidation at the terminal carbon of the ethyl group on the diazepinone moiety.
M17.
This metabolite had a protonated molecular ion at m/z 428, corresponding to a loss of 14 amu from BILR 516. The product ion spectrum of M17 showed abundant product ions at m/z 239 and 267 (Fig. 2D), which were also 14 amu less than the corresponding product ions of BILR 516. Thus, it is likely that M17 is formed after demethylation on the diazepinone ring from BILR 516.
The retention times and characteristic product ions of the parent and all metabolites are listed in Table 2. In addition to BILR 516, standards of nine metabolites, including INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, BILR 402, BI 212580 (M14), and BI 212578 (M17), were also synthesized on the basis of proposed structures. The 1H and 13C NMR data of the parent and synthetic standards of the metabolites are listed in supplemental data. The structures of the 10 metabolites were confirmed by comparison with synthetic standards based on the same retention times, molecular weight, and MS/MS patterns. The overall metabolic pathways of BILR 355 are proposed in Fig. 3. The names of the standards are also listed in the final column in Table 2 and next to the metabolite names in Fig. 3.

Proposed metabolite structures and metabolic pathways of BILR 355 in humans.
Quantitation of Metabolites of BILR 355 in Clinical Samples.
The levels of the nine metabolites with authentic standards, including INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, BILR 516, BI 212580, and BI 212578, were measured in clinical samples from individual subjects after the last dose of BILR 355 and RTV on day 7 (steady state). The quantitation result showed that INLA 1678, BI 211136, BIRK 122, EXLS 1451, INRF 105, BILR 564, BI 212580, and BI 212578 were minor metabolites of BILR 355, with plasma concentration of the tested metabolites less than 0.22 μM in all clinical samples. BILR 402 was also monitored in a separate study and has been shown to be a minor metabolite of BILR 355 with Cmax of 0.18 μM (Boehringer Ingelheim Pharmaceuticals, Inc., data on file).
The levels of BILR 516 were surprisingly high at steady state, exceeding levels of the parent drug. The mean plasma concentration-time profiles of BILR 516 on day 1 and steady state are plotted in Fig. 4. The BILR 355 profiles are also shown in Fig. 4 for comparison purposes (Huang et al., 2009). The calculated Cmax, area under the plasma concentration-time curve over the dosing interval (AUC0–τ), and t1/2 of BILR 516 and BI 212578, and comparison of these values with those obtained for BILR 355 (Huang et al., 2009) are shown in Table 3.

Plasma concentration-time profiles of BILR 355 and BILR 516 on day 1 and day 7 (steady state) in humans.
Pharmacokinetic parameters of BILR 355, BILR 516, and BI 212578 after 7-day dosing of BILR 355 (150 mg)/RTV (100 mg) (b.i.d.) in humans
Data are means ± S.D. or percentages for BILR 516/BILR 355 and BI 212578/BILR 355 ratios.
Discussion
BILR355 is a second-generation NNRTI developed to treat HIV-1 infection. BILR 355 has been shown to be a good substrate of CYP3A with a relatively low apparent Km of 0.79 μM and a high turnover in HLMs. Extensive first-pass metabolism by intestinal and hepatic CYP3A was proposed as an explanation for the low systemic exposure and short half-life after oral administration to humans (Huang et al., 2008). A favorable PK profile, with increased systemic exposure, was achieved after concomitant administration of the CYP3A inhibitor RTV (Huang et al., 2008) as seen with other HIV drugs (Hull and Montaner, 2011). An unexpected consequence of the inhibition of this primary clearance pathway was a metabolic switching, leading to the formation of a disproportionate human metabolite.
First, the in vitro metabolism of BILR 355 was evaluated in HLMs. As summarized in Table 1, the calculated intrinsic clearance of BILR 355 by HLMs appears to be fairly high. The average CLh/Qh from four HLM lots was 82%, indicating that BILR 355 may go through extensive phase I metabolism in vivo. The apparent Km values for BILR 355 metabolism ranged from 0.63 to 1.14 μM with a mean value of 0.79 μM. In the clinical phase Ia study with administration of single doses of BILR 355 from 12.5 to 100 mg, Cmax ranged from 14.8 ng/ml (0.03 μM) to 937 ng/ml (2.12 μM) (Huang et al., 2008), which bracketed the range of apparent Km values shown above. Therefore, it was not surprising to see the mean exposure (AUC0–∞) increase superproportionally (71.8–1310 ng · h/ml) and the mean apparent clearance (CL/F) decrease (246–79.2 l/h) over this range of doses in the phase Ia study (Huang et al., 2008). The observed nonlinearity in AUC0–∞ and CL/F is probably due to saturation of the metabolism of BILR 355.
CYP3A4 was identified as the major P450 responsible for the metabolism of BILR 355, of the six major human P450 isoforms tested, as only rCYP3A4 caused a detectable depletion of BILR 355 and only ketoconazole (CYP3A4 specific inhibitor) resulted in significant inhibition of the metabolism of BILR 355 by HLMs (complete inhibition with 3 μM ketoconazole). In addition, the apparent Vmax values of BILR 355 metabolism correlated well with the reported CYP3A4 activity of the different lots of HLMs, but no correlation was observed with activities of CYP1A2, CYP2A6, CYP2C19, or CYP2D6. The depletion rates of BILR 355 in the presence of rCYP3A4 were similar at substrate concentrations of 1 and 10 μM. This finding suggests that the apparent Km for the CYP3A4-catalyzed metabolism of BILR 355 is less than 1 μM, which is consistent with the apparent Km values for BILR 355 metabolism in HLMs. Overall, the combination of the results from recombinant P450 profiling, chemical inhibition, and correlation analysis supports the fact that CYP3A4 plays an important role in the metabolism of BILR 355. RTV, a potent CYP3A inhibitor, was therefore used as a boosting agent for BILR 355 and effectively elevated the exposure of BILR 355 in humans (Huang et al., 2008), which further confirms that BILR 355 is extensively metabolized by CYP3A. The involvement of CYP3A5 was not specifically tested in these studies. However, because there is typically an overlap in substrate specificity between CYP3A4 and CYP3A5, it is possible that CYP3A5 is also involved in the metabolism of BILR 355. Overall, the current phenotyping results support the concept that the increase in BILR 355 systemic exposure with concomitant administration of RTV is primarily due to inhibition of CYP3A.
It was expected that the levels of metabolites would decrease with concomitant administration of the CYP3A inhibitor RTV. Metabolite profiling studies for the phase Ib plasma samples, in which RTV was administered with BILR 355, showed a total of 18 metabolites. The structures of 10 of these metabolites were confirmed on the basis of comparison with authentic standards. Although almost all of these metabolites were minor on the basis of quantitation using authentic standards or peak area estimations for metabolites without authentic standards, there was one metabolite found at substantial levels, BILR 516. This metabolite was not observed in human plasma when BILR 355 was administered alone (Boehringer Ingelheim Pharmaceuticals, Inc., data on file).
BILR 516 accumulated in humans over time (Fig. 4), which was attributable to its long t1/2 (54.5 h) (Table 3). The t1/2 of BILR 516 is significantly longer than the t1/2 of the parent compound (14.7 h) (Table 3). As a result, the exposure of BILR 516 was even higher than the exposure of the parent drug at steady state (Fig. 4). Because the exposure of BILR 516 was greater than 10% of the total drug-related materials, it was identified as a major human metabolite (U.S. Food and Drug Administration, 2008).
On the basis of both the U.S. Food and Drug Administration guidance and the later published 2009 ICH guidance M3 (R2) (U.S. Food and Drug Administration, 2008; European Medicines Agency, 2009), the coverage of BILR 516 in toxicology species needed to be assessed. Additional studies showed that the levels of BILR 516 were very low in Sprague-Dawley rats and beagle dogs, the two general toxicology species for BILR 355 (Boehringer Ingelheim Pharmaceuticals, Inc., data on file). Therefore, BILR 516 was defined as a disproportionate human metabolite. A better understanding of the formation of BILR 516 was needed to help select an appropriate toxicology species (Li et al., 2012).
Likewise, BI 212580 and BI 212578 have the same quinolone structure as BILR 516. It is possible that both metabolites may have longer half-lives similar to that of BILR 516 with a potential to accumulate in the body over time and become major metabolites. Therefore, both metabolites were synthesized on the basis of the proposed structures, and their exposure was assessed using the authentic standards and compared with the exposure of the parent. The pharmacokinetic parameters of BI 212578 are included in Table 3. The concentrations of BI 212580 were lower than the limit of quantitation (0.0426 μM) at most time points, and thus its pharmacokinetic parameters could not be calculated. BI 212578 does have a long t1/2 of 33 h, as anticipated. Both BI 212580 and BI 212578 were confirmed as minor metabolites, and no further assessment was warranted.
Overall, the exposure of BILR 355 was significantly improved by concomitant administration of RTV, mainly due to suppression of CYP3A-mediated metabolism of BILR 355. However, as a consequence, a disproportionate human metabolite, BILR 516, emerged, which was not detected in human plasma samples when BILR 355 was administered alone. The resulting consequence of a new major metabolite posed several challenges for drug development. This article raises a concern about potential metabolic switching with concomitant administration of a potent boosting agent or any potent inhibitors of major metabolic pathways. The metabolic pathways for the formation of BILR 516 from BILR 355 turned out to be complicated as outlined in the accompanying publication (Li et al., 2012). In brief, BILR 516 is formed by sequential reactions mediated by two non-P450 enzymes. First, BILR 355 is transformed to BILR 402 by gut bacteria through reduction of the N-oxide and then BILR 402 is metabolized to BILR 516 by aldehyde oxidase.
Authorship Contributions
Participated in research design: Li, Lai, and Tweedie.
Conducted experiments: Li, Lai, and Whitcher-Johnstone.
Contributed new reagents or analytic tools: Busacca, Eriksson, and Lorenz.
Performed data analysis: Li and Lai.
Wrote or contributed to the writing of the manuscript: Li and Tweedie.
Acknowledgments
We thank Dr. Fenglei Huang for providing the Cmax data of BILR 402 and thank Dr. Timothy S. Tracy for scientific advice and review of the manuscript.
Footnotes
This research was funded by Boehringer Ingelheim Pharmaceuticals, Inc.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- HIV-1
- human immunodeficiency virus
- NNRTI
- non-nucleoside reverse transcriptase inhibitor
- BILR 355
- 11-ethyl-5,11-dihydro-5-methyl-8-[2-[(1-oxido-4-quinolinyl)oxy]ethyl]-6H-dipyrido[3,2-b:2′,3′-e][1,4]diazepin-6-one
- P450
- cytochrome P450
- RTV
- ritonavir
- AUC
- area under the plasma concentration-time curve
- HLM
- human liver microsomes
- r
- recombinant
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- PK
- pharmacokinetic
- HPLC
- high-performance liquid chromatography
- amu
- atomic mass units.

- Received December 16, 2011.
- Accepted March 5, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}