


Abstract
Esterases catalyze the hydrolysis of therapeutic drugs containing esters or amides in their structures. Human carboxylesterase (CES) and arylacetamide deacetylase (AADAC) are the major enzymes that catalyze the hydrolysis of drugs in the liver. Characterization of the enzyme(s) responsible for drug metabolism is required in drug development and to realize optimal drug therapy. Because multiple enzymes may show a metabolic potency for a given compound, inhibition studies using chemical inhibitors are useful tools to determine the contribution of each enzyme in human tissue preparations. The purpose of this study was to find specific inhibitors for human CES1, CES2, and AADAC. We screened 542 chemicals for the inhibition potency toward hydrolase activities of p-nitrophenyl acetate by recombinant CES1, CES2, and AADAC. We found that digitonin and telmisartan specifically inhibited CES1 and CES2 enzyme activity, respectively. Vinblastine potently inhibited both AADAC and CES2, but no specific inhibitor of AADAC was found. The inhibitory potency and specificity of these compounds were also evaluated by monitoring the effects on hydrolase activity of probe compounds of each enzyme (CES1: lidocaine, CES2: CPT-11, AADAC: phenacetin) in human liver microsomes. Telmisartan and vinblastine strongly inhibited the hydrolysis of CPT-11 and/or phenacetin, but digitonin did not strongly inhibit the hydrolysis of lidocaine, indicating that the inhibitory potency of digitonin was different between recombinant CES1 and liver microsomes. Although we could not find a specific inhibitor of AADAC, the combined use of telmisartan and vinblastine could predict the responsibility of human AADAC to drug hydrolysis.
Introduction
Drug-metabolizing enzymes play pivotal roles in the detoxification, elimination, or pharmacological activation of drugs. Characterization of the enzyme(s) responsible for drug metabolism is required for the development of new drugs and the realization of optimal drug therapy. Esterases help metabolize 10% of clinical drugs to either detoxify or activate them (Fukami and Yokoi, 2012). Among esterases involved in drug metabolism, carboxylesterase (CES) has been well studied. Human CES enzymes are classified into five subfamilies: CES1, CES2, CES3, CES4A, and CES5A (Holmes et al., 2010). CES1 (EC3.1.1.1, EC3.1.1.56) and CES2 (EC3.1.1.1, EC3.1.1.56, EC3.1.1.84) are mainly responsible for the biotransformation of a number of clinically used drugs. These esterases are commonly expressed in the liver, which is the main organ responsible for drug metabolism (Fukami and Yokoi, 2012). CES enzymes are mainly localized on the lumen side of the endoplasmic reticulum, but they are also present in human liver cytosol with variable expression levels among individuals (Xu et al., 2002; Fukami et al., 2008). Recently, we found that arylacetamide deacetylase (AADAC) (EC3.1.1.3), which is also localized on the lumen side of the ER (Frick et al., 2004), is involved in the hydrolysis of some clinical drugs such as flutamide, phenacetin, and rifamycins (Watanabe et al., 2009, 2010; Nakajima et al., 2011). Importantly, the hydrolysis of flutamide and phenacetin by AADAC is linked with hepatotoxicity and nephrotoxicity or hematic toxicity, respectively. With accumulating evidence of AADAC function in the formation of toxic metabolites or in detoxification, AADAC should be prioritized as an enzyme capable of catalyzing the hydrolysis of clinically used drugs.
Another esterase family is paraoxonase (PON) enzymes, which are also expressed in the endoplasmic reticulum membrane in liver cells (Gonzalvo et al., 1998). PON enzymes are highly present in human plasma (Ng et al., 2001), and therefore, their roles in drug metabolism within the plasma have been well studied. However, little is known about the roles of PON enzymes in the liver. PON enzymes require calcium to exert their activities (Kuo and La Du, 1998). The drug-metabolizing activities of PON enzymes in the liver cannot be determined using in vitro studies on liver preparations because calcium chloride is not usually added to the incubation mixture. Butyrylcholinesterase exists in human plasma. Jbilo et al. (1994) reported that butyrylcholinesterase mRNA was detected in human liver, but the enzyme activity appeared to be undetectable in the liver as well as in intestinal fractions (Takusagawa et al., 2012). Taken together, if a drug is found to be hydrolyzed by human liver microsomes, the enzymes responsible for the hydrolysis are likely CES or AADAC. The identification of enzymes involved in drug metabolism is an important step in drug development and is useful for the prediction of drug-drug interactions.
Recombinant enzymes are used to investigate whether an endogenous enzyme has the ability to metabolize a given compound. However, if several enzymes show a metabolic potency, their relative contribution in human tissue should be determined. A representative method for this evaluation is inhibitory analysis. Inhibition studies use specific inhibitors of each enzyme to determine the enzyme responsible for drug metabolism. Bis-(4-nitrophenyl) phosphate (BNPP) and diisopropyl fluorophosphate are often used as CES inhibitors (Heymann and Krisch, 1967; Yamaori et al., 2006), but they also inhibit AADAC (Watanabe et al., 2009) (The abbreviation for BNPP in our previous paper, Watanabe et al., 2009, was incorrect. It was corrected as an erratum). Eserine, a known cholinesterase inhibitor (Iwatsubo, 1965), strongly inhibits CES2 and AADAC (Kobayashi et al., 2012a). Thus, inhibitors specific for either CES or AADAC remain to be found. In the present study, we sought to discover specific inhibitors of human CES1, CES2, and AADAC.
Materials and Methods
Chemicals and Reagents.
Lidocaine, p-nitrophenol (PNP), phenacetin, and 2,6-xylidine were purchased from Wako Pure Chemical Industries (Osaka, Japan). p-Nitrophenyl acetate (PNPA) and p-phenetidine were purchased from Sigma-Aldrich (St. Louis, MO). Irinotecan hydrochloride (CPT-11) and 7-ethyl-10-hydroxycamptothecin (SN-38) were purchased from Toronto Research Chemicals (Ontario, Canada). The suppliers of the 542 compounds used as inhibitors in this study are shown in Supplemental Table 1. All other chemicals were of analytical grade or the highest quality commercially available.
PNPA Hydrolase Activities.
The hydrolase activity of PNPA was determined as described previously using a substrate concentration of 100 µM (Shimizu et al., 2012). A typical incubation mixture (at a final volume of 0.2 ml) contained 100 mM potassium phosphate buffer (pH 7.4); homogenates of Sf21 cells expressing CES1 (NP_001020365.1), CES2 (NP_003860.2), or AADAC (NP_001077.2) (0.1 mg/ml), which were constructed in our previous studies (Fukami et al., 2010; Watanabe et al., 2010); and the inhibitor candidates shown in Supplemental Table 1. The concentrations of the inhibitor candidates were 100 µM (Supplemental Table 1) unless otherwise noted. Because of the limited solubility, the concentration of ethizolam, fenbendazole, and telmisartan was at most 50 µM. To determine the concentrations that caused 50% inhibition (IC50), the various concentrations of digitonin, telmisartan, and vinblastine were used in the ranges of 0.1–100 µM, 0.1–50 µM, and 0.1–100 µM, respectively. The organic solvents used to dissolve the inhibitors are shown in Supplemental Table 1. The final concentration of the organic solvent in the incubation mixture was 1.5% (for telmisartan, 5%). The control activity was measured with the corresponding concentration of the organic solvents. The reaction was initiated by the addition of PNPA after a 2-minute preincubation at 37°C. The preincubation time referred to previous reports (Karanth and Pope, 2000; Takusagawa et al., 2012; Yanjiao et al., 2013; Ohura et al., 2014). After a 1-minute incubation at 37°C, the reaction was terminated by the addition of 100 µl of ice-cold methanol. It was confirmed that the rate of PNP formation was linear in conditions with protein concentrations of up to 0.4 mg/ml and incubation times up to 3 minutes and that 50% methanol in a final volume could stop the hydrolytic reaction (data not shown). The formed PNP was measured by absorbance at 405 nm using a Biotrak II plate reader (GE Health Science). The quantification of PNP was determined by comparing the absorbance with that of an authentic standard. Because PNP contaminants exist in the commercially available PNPA to the extent of ∼5%, the content of PNP in the control mixture incubated without the enzyme was subtracted from that in the mixture with the enzyme. IC50 values were calculated using the equation as follows:

where A and B are the higher and lower concentrations near 50% inhibition, respectively, and C and D are the inhibition percentages at B and A, respectively.
Lidocaine, CPT-11, and Phenacetin Hydrolase Activities.
The hydrolysis of lidocaine, CPT-11, and phenacetin was used to probe the activities of CES1, CES2, and AADAC, respectively, according to our previous studies (Fukami et al., 2010; Watanabe et al., 2010; Higuchi et al., 2013). A typical incubation mixture (at a final volume of 0.2 ml) contained 100 mM potassium phosphate buffer (pH 7.4), human liver microsomes (HLM) (pooled, n = 50, BD Gentest, Woburn, MA), or the homogenates of Sf21 cells expressing CES1, CES2, and AADAC (0.1 mg/ml). The hydrolysis of lidocaine was measured at a concentration of 500 µM and a protein concentration of 0.3 mg/ml. The hydrolysis of CPT-11 was measured at a concentration of 2 µM and a protein concentration of 0.2 mg/ml. The hydrolysis of phenacetin was measured at a substrate concentration of 1 mM and a protein concentration of 0.4 mg/ml. Digitonin (0–100 µM), telmisartan (0–50 µM), and vinblastine (0–100 µM) were added as inhibitors.
To determine the Ki (inhibition constant) values for the CPT-11 hydrolase activities, CPT-11 was added in concentrations ranging from 2 to 10 µM, and the inhibitors telmisartan and vinblastine were added in concentrations ranging from 0 to 5 µM and 0 to 80 µM, respectively. For determination of the Ki values for the phenacetin hydrolase activities, phenacetin was added in concentrations ranging from 0.5 to 4 mM, and the inhibitor vinblastine was added in concentrations ranging from 0 to 10 µM. The Ki values and inhibition types were determined by fitting the kinetic data to a competitive, noncompetitive, uncompetitive, or mixed inhibition model by nonlinear regression analysis using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Results and Discussion
Known Inhibitors for Human CES1 and CES2.
We previously reported that human AADAC hydrolyzes flutamide (Watanabe et al., 2009), phenacetin (Watanabe et al., 2010), rifamycins (Nakajima et al., 2011), and PNPA (Watanabe et al., 2009). PNPA is a known marker of CES activity (Hosokawa et al., 1995). Flutamide is also hydrolyzed by CES2 (Kobayashi et al., 2012b). These findings revealed that substrate specificity of AADAC appears to partly overlap with those of CES enzymes. Inhibition studies using specific inhibitors of certain enzymes can be used to identify the enzymes responsible for drug metabolism or to determine the contribution of each enzyme. Several compounds have been reported to be potent inhibitors of human CES enzymes. For example, benzil analogs potently inhibit both CES1 and CES2 (Wadkins et al., 2005, Parkinson et al., 2011). Trifluoromethylketone-containing compounds also have potent inhibitory effects on both CES enzymes (Wadkins et al., 2007). Sulfonamide analogs seem to preferentially inhibit CES2 enzyme activity rather than CES1 enzyme activity (Wadkins et al., 2004). Loperamide has been frequently used as a human CES2 inhibitor (Quinney et al., 2005). It should be noted that these compounds have never been investigated for the potential of AADAC inhibition. In addition, no studies have been performed to compare the inhibition potency of compounds toward CES and AADAC. According to the background, we sought to find specific inhibitors of human CES and AADAC by screening commercially available drugs or compounds.
Screening of Inhibitors of CES and AADAC Enzymes by Measuring PNPA Hydrolysis.
The inhibitory effects of 542 compounds on the hydrolysis of PNPA by recombinant CES and AADAC enzymes were investigated (Supplemental Table 2). The control activities of recombinant CES1, CES2, and AADAC were 167.0, 289.9, and 244.4 nmol/min/mg protein, respectively. Inasmuch as the activity by mock-transfected Sf21 cell homogenates was marginal (13.4 nmol/min/mg protein), the residual activities in the presence of inhibitor candidates were calculated as relative to above control activities. In this study, candidate inhibitors were also preincubated because of a way frequently used. Compounds showing 80% or more inhibition were defined as strong inhibitors, whereas compounds showing 15% or less inhibition were defined as noninhibitors. Eighteen compounds were categorized as strong inhibitors for each esterase (Table 1). Among them, 11 compounds showed strong inhibition toward CES1, CES2, and AADAC. We previously reported that nordihydroguaiaretic acid and simvastatin strongly inhibit both CES1 and CES2 enzyme activities (Takahashi et al., 2009; Fukami et al., 2010). The present study demonstrated that these compounds also strongly inhibited AADAC. Interestingly, there is no commonality in the chemical structure between the 11 compounds. Although we did not examine further for these compounds in this study, these compounds may reveal different inhibitory potency between enzymes at concentrations lower than 100 µM. Such study may be worth pursuing in the future.
Compounds showing strong inhibitory effects on PNPA hydrolysis by recombinant CES1, CES2, and AADAC
PNPA hydrolysis by recombinant CES1, CES2, and AADAC was determined at a substrate concentration of 100 μM. The control activity values of recombinant CES1, CES2, and AADAC were 167.0, 289.9, and 244.4 nmol/min/mg protein, respectively.
2-Chloro-3′,4′-dimethoxybenzil, phenylmethylsulfonyl fluoride (PMSF), and JW480 strongly inhibited the activities of both CES1 and CES2 but not AADAC (Table 1). It was originally reported that various benzil analogs could potently inhibit o-nitrophenyl acetate hydrolysis by CES1 and CES2 (Wadkins et al., 2005; Parkinson et al., 2011). However, the present study found that benzil could not inhibit AADAC enzyme activity. We previously reported that PMSF weakly inhibited the hydrolysis of flutamide and rifamycins by AADAC (Watanabe et al., 2009; Nakajima et al., 2011), suggesting that the marginal inhibitory effect of PMSF on AADAC would be independent of substrates. Xie et al. (2002) reported that 100 µM PMSF potently inhibited the hydrolase activity of 4-nitrophenyl acetate by CES1 and CES2 to <10% of residual activity, supporting our results (Table 1). JW480 is a known inhibitor of neutral cholesterol ester hydrolase 1, also known as arylacetamide deacetylase-like 1 (AADACL1) (Chang et al., 2011). Although we expected that JW480 would potently inhibit AADAC enzyme activity because AADACL1 shares moderate homology with AADAC except for in the N-terminal region, its inhibitory potential was marginal.
Digitonin specifically inhibited CES1. To our knowledge, this is the first report of this compound as a specific inhibitor of CES1. We further evaluated the inhibition potency and specificity of digitonin using HLM as an enzyme source.
Flavoxate and telmisartan specifically inhibited CES2. When flavoxate was added to the incubation mixture, approximately half of it was nonenzymatically converted to 3-methylflavone-8-carboxylic acid (data not shown), which inhibited AADAC, CES1, and CES2 enzyme activities by 37, 70, and 71%, respectively (Supplemental Table 2). Because the extent of the conversion may influence the inhibition potency, flavoxate may not be a user friendly inhibitor. Telmisartan would be a better CES2-specific inhibitor.
Vinblastine strongly inhibited both CES2 and AADAC but did not inhibit CES1. Unfortunately, strong and specific inhibitors of AADAC were not found. However, the combined use of telmisartan and vinblastine would predict the responsibility of AADAC in the hydrolysis of drugs.
PNPA hydrolase activities were enhanced by some compounds such as bilirubin (% of control, CES1: 350%, CES2: 145%), N-cyclohexyl-3-aminopropanesulfonic acid (AADAC: 169%), and 1-decanesulfonic acid (CES2: 230%, AADAC: 119%) (Supplemental Table 2). As for AADAC, we previously found that phenacetin hydrolase activity was enhanced by flutamide (Watanabe et al., 2010). As for CES1 and CES2, there were no experimental reports for such enhancement of activity by compounds. However, it has been reported that CES1 has three ligand-binding sites, an active site, a side door, and a Z-site, and the Z-site plays a role in the allosteric activation of catalysis (Bencharit et al., 2006). Our study could prove the allosteric activation of CES1. Because we found the allosteric activation of CES2, the results suggest the possibility that CES2 and AADAC have multiple binding sites.
IC50 Values of Digitonin, Telmisartan, and Vinblastine for PNPA Hydrolysis by Esterases.
We determined the IC50 values of digitonin, telmisartan, and vinblastine for CES1, CES2, and AADAC activities (Fig. 1). For comparison, the IC50 value of loperamide (a known CES2 inhibitor) (Quinney et al., 2005) was also determined. The IC50 value of digitonin for CES1 enzyme activity was 9.2 ± 0.4 µM. The IC50 value of telmisartan for CES2 activity was 0.5 ± 0.05 µM. Loperamide inhibited CES2 activity with a lower IC50 value (0.1 ± 0.003 µM) than telmisartan, but it moderately inhibited CES1 and AADAC activities with IC50 values of over 100 µM. Accordingly, telmisartan would be a more appropriate specific inhibitor of CES2. It was reported that loperamide potently inhibited the hydrolase activities of 4-methylumbilliferyl acetate at 0.2 mM substrate concentration and PNPA at 1 mM substrate concentration by CES2 with 0.38 µM (Quinney et al., 2005) and 1.12 µM (Wang et al., 2011) of IC50 values, respectively, values that were higher than obtained in this study (0.1 ± 0.003 µM). However, the fact remains that loperamide can potently inhibit CES2 enzyme activity. The IC50 values of vinblastine for CES2 and AADAC enzyme activities were 3.6 ± 0.2 and 9.4 ± 0.6 µM, respectively.
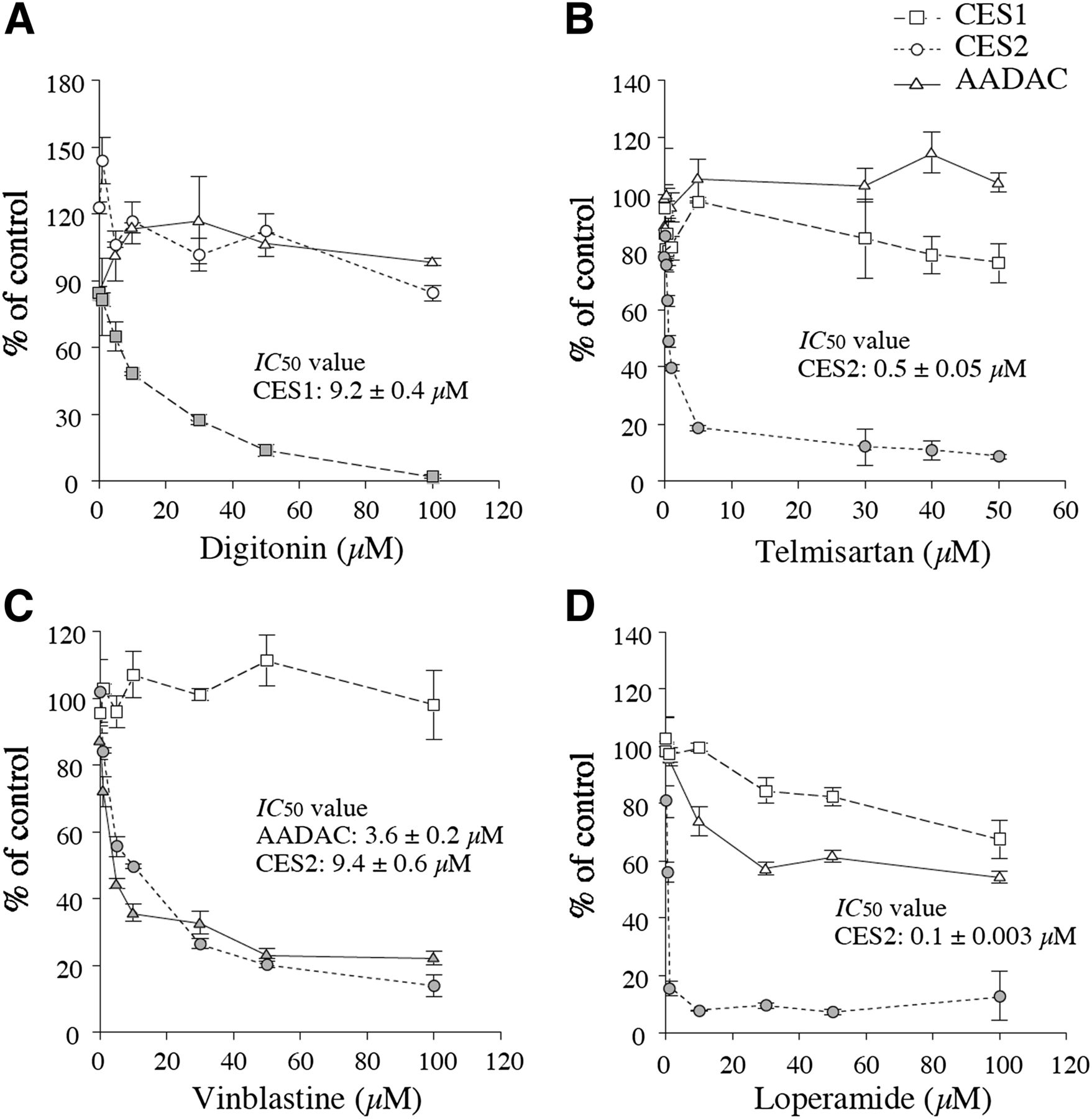
Inhibitory effects of digitonin (A), telmisartan (B), vinblastine (C), and loperamide (D) on PNPA hydrolysis by recombinant AADAC, CES1, and CES2. The PNPA concentration was 100 μM. The control activities of recombinant CES1, CES2, and AADAC were 167.0, 289.9, and 244.4 nmol/min/mg protein, respectively. Each point represents the mean ± S.D. of triplicate determinations.
Inhibition Potencies of Digitonin for Probe Activities of Each Esterase.
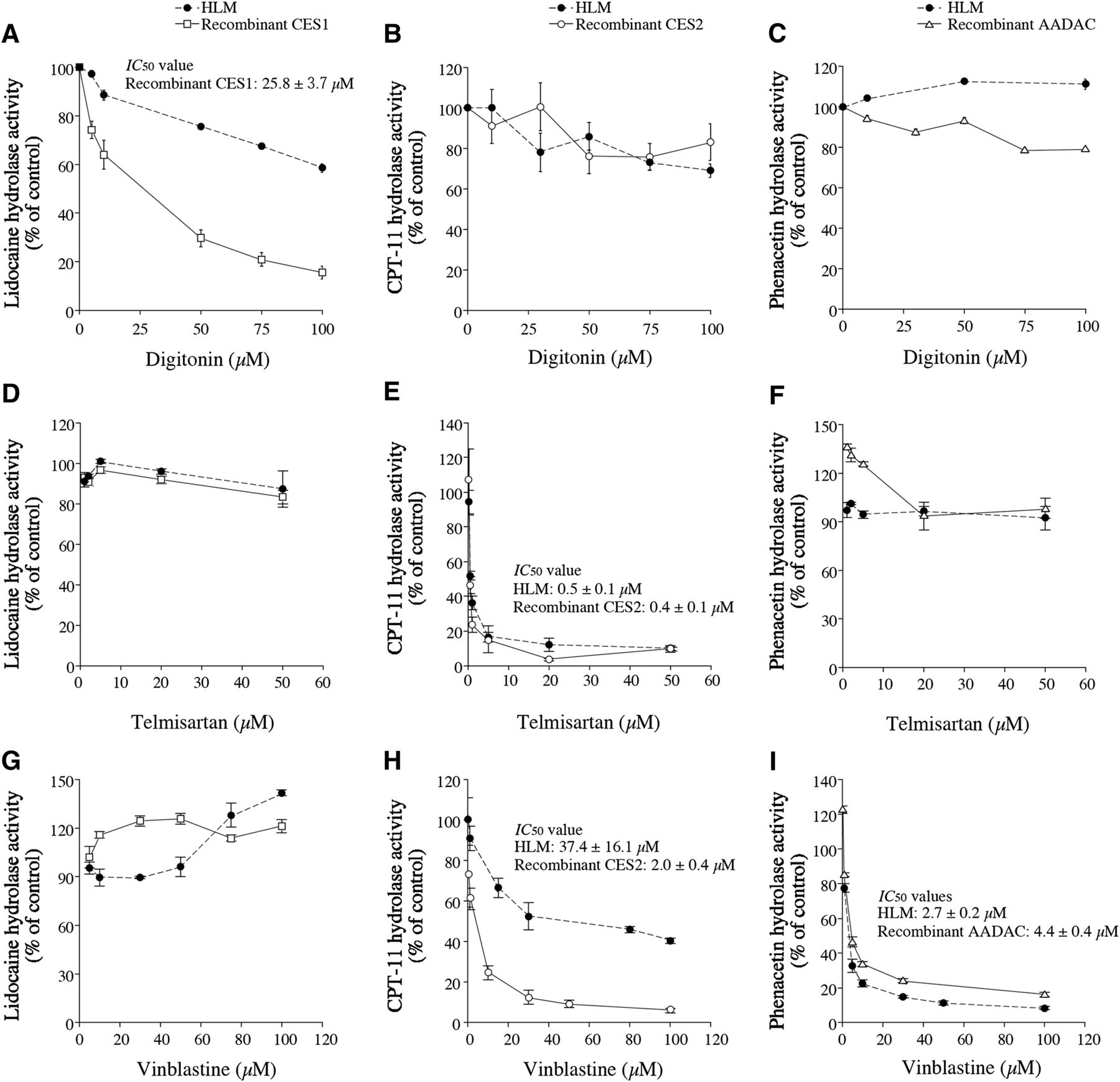
The inhibition selectivity of digitonin toward CES1 was evaluated by measuring the hydrolysis of lidocaine, CPT-11, and phenacetin, which are probes for the activities of CES1, CES2, and AADAC, respectively, by HLM and recombinant esterases (Fig. 2, A–C). Digitonin inhibited the hydrolysis of lidocaine by recombinant CES1 (IC50 = 25.8 ± 3.7 µM); however, it did not potently inhibit the activity in HLM, and thus, IC50 value was not calculated (Fig. 2A). The weak inhibitory potency of digitonin toward lidocaine hydrolysis in HLM was also observed for fenofibrate hydrolysis (Supplemental Fig. 1), which is also specifically catalyzed by CES1 (Fukami et al., 2010). We previously reported that the IC50 values of diisopropyl fluorophosphate and BNPP on the hydrolysis of capecitabine, which is specifically catalyzed by CES1, in HLM were approximately 10-fold higher than those by purified CES1 (Tabata et al., 2004). The Ki value of troglitazone for imidapril hydrolysis, which is also specifically catalyzed by CES1, in HLM was approximately ninefold higher than that of recombinant CES1 (Fukami et al., 2010). The weak inhibitory potency of digitonin toward CES1 in HLM might be caused by the interference of some proteins in HLM. Digitonin slightly inhibited the hydrolysis of CPT-11 and phenacetin with either HLM or recombinant enzymes, although the IC50 values could not be calculated (Fig. 2, B and C). Taken together, we found that digitonin is a specific inhibitor of CES1, but the inhibitory potency for HLM was mild.

Inhibitory effects of digitonin (A, B, C), telmisartan (D, E, F), and vinblastine (G, H, I) on lidocaine (A, D, G), CPT-11 (B, E, H), and phenacetin (C, F, I) hydrolysis by HLM and recombinant enzymes. The IC50 values were calculated by evaluating the hydrolysis of lidocaine (500 µM), CPT-11 (2 µM), and phenacetin (1 mM), which were used for probe substrates of CES1, CES2, and AADAC, respectively. The control lidocaine hydrolysis values of recombinant CES1 and HLM were 23.2 and 154.6 pmol/min/mg protein, respectively. The control CPT-11 hydrolysis values by recombinant CES2 and HLM were 1.8 and 1.4 pmol/min/mg protein, respectively. The control phenacetin hydrolysis values by recombinant AADAC and HLM were 1.1 and 0.8 nmol/min/mg protein, respectively. Each point represents the mean ± S.D. of triplicate determinations.
Inhibition Potencies of Telmisartan for Probe Activities of Each Esterase.
The inhibition selectivity of telmisartan toward CES2 was evaluated by measuring the hydrolysis of lidocaine, CPT-11, and phenacetin by HLM and recombinant esterases (Fig. 2, D–F). Telmisartan inhibited the hydrolysis of CPT-11 by recombinant CES2 (IC50 = 0.4 ± 0.1 µM) and HLM (IC50 = 0.5 ± 0.1 µM) (Fig. 2F) but not the hydrolysis of lidocaine and phenacetin (Fig. 2, D and E). The inhibitory constant (Ki) of telmisartan for CPT-11 hydrolysis by HLM was evaluated and was found to be 1.2 ± 0.1 µM with a noncompetitive-type inhibition (Fig. 3A). Telmisartan could be a useful specific inhibitor of CES2.

Inhibitory effects of telmisartan and vinblastine on CES2 and CES2/AADAC enzyme activities in HLM. (A) The Ki value of telmisartan on CPT-11 hydrolysis by HLM was calculated. The substrate concentrations of CPT-11 ranged from 2 to 10 µM. The Ki values of vinblastine on CPT-11 (B) and phenacetin (C) hydrolysis by HLM were calculated. The substrate concentrations of CPT-11 and phenacetin ranged from 2 to 10 µM and 0.5 to 4 mM, respectively. Each point represents the mean ± S.D. of triplicate determinations. 1/V, 1/velocity.
Inhibition Potencies of Vinblastine for Probe Activities of Each Esterase.
The inhibition selectivity of vinblastine toward CES2 and AADAC was evaluated by measuring the hydrolysis of lidocaine, CPT-11, and phenacetin by HLM and recombinant esterases (Fig. 2, G-I). Vinblastine inhibited the hydrolysis of CPT-11 by recombinant CES2 (IC50 = 2.0 ± 0.4 µM) and HLM (IC50 = 37.4 ± 16.1 µM) and the hydrolysis of phenacetin by recombinant AADAC (IC50 = 4.4 ± 0.4 µM) and HLM (IC50 = 2.7 ± 0.2 µM) (Fig. 2, H and I) but not lidocaine hydrolysis (Fig. 2G). The IC50 value of vinblastine for CPT-11 hydrolysis in HLM was higher than that in recombinant CES2 (Fig. 2H). Humerickhouse et al. (2000) reported that CES1 also hydrolyzes CPT-11, although the Km value (43 µM) is 10-fold higher than that of CES2 (3.4 µM). The dissociation of the IC50 values between HLM and recombinant CES2 implies a possibility that CES1 might partly contribute to CPT-11 hydrolysis in the human liver, because CES1 is more predominantly expressed in the liver than is CES2 (Sato et al., 2012), even at a low substrate concentration (2 µM). The Ki value of vinblastine for CPT-11 hydrolysis in HLM was evaluated and was found to be 66.1 ± 6.1 µM with a noncompetitive-type inhibition (Fig. 3B). The Ki value for phenacetin hydrolysis by HLM was also evaluated and was found to be 2.4 ± 0.2 µM with a mixed-type inhibition (Fig. 3C). Taken together, vinblastine could be useful as an inhibitor of CES2 and AADAC and combined use with telmisartan may predict the responsibility of AADAC in drug hydrolysis.
Prediction for the Effect of Telmisartan and Vinblastine on CES2 and AADAC Enzyme Activity in Clinical Use.
Because we found that telmisartan and vinblastine potently inhibit CES2 and CES2/AADAC, respectively, one may be concerned with drug-drug interactions (DDIs). The maximum unbound concentration of telmisartan at the inlet to the liver is estimated to be 0.12 µM after oral administration of the clinical dose of 160 mg (Ishiguro et al., 2006). The Ki value of telmisartan for CPT-11 hydrolysis by HLM (1.2 ± 0.1 µM) was 10-fold higher than the estimated concentration of telmisartan in the liver. In contrast, the concentration of telmisartan in the intestine in which CES2 is highly expressed was predicted to be 31 µM (Nakamori et al., 2012). Thus, telmisartan may exhibit a DDI with compounds that are substrates of CES2 via inhibition of CES2 in the intestine but not in the liver. The plasma concentration of vinblastine within 96 hours after a 24-hour continuous intravenous infusion (1.5 mg/m2) was estimated to be below 6 nM (Stewart et al., 1983), which is far lower than the Ki values for CPT-11 and phenacetin hydrolysis by HLM. Therefore, the inhibitory effect of vinblastine on AADAC and CES2 enzyme activities is unlikely to lead to critical DDIs.
Conclusion
We found that digitonin specifically inhibited CES1 activity, telmisartan specifically inhibited CES2 activity, and vinblastine inhibited CES2 and AADAC activities. The combined use of vinblastine and telmisartan may lead to the clarification of AADAC involvement.
Authorship Contributions
Participated in research design: Shimizu, Fukami, Nakajima, and Yokoi.
Conducted experiments: Shimizu and Fukami.
Contributed new reagents or analytic tools: Shimizu.
Performed data analysis: Shimizu and Fukami.
Wrote or contributed to the writing of the manuscript: Shimizu, Fukami, Nakajima, and Yokoi.
Footnotes
- Received January 8, 2014.
- Accepted April 21, 2014.
This work was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science [23590174].
Abbreviations
- AADAC
- arylacetamide deacetylase
- BNPP
- bis-(4-nitrophenyl) phosphate
- CES
- carboxylesterase
- DDI
- drug-drug interactions
- HLM
- human liver microsomes
- PMSF
- phenylmethylsulfonyl fluoride
- PNPA
- p-nitrophenyl acetate
- PON
- paraoxonase
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}