


Abstract
We developed murine Cyp3a knockout (KO) chimeric mice with humanized liver expressing human P450s similar to those in humans and whose livers and small intestines do not express murine Cyp3a. This approach may overcome effects of residual mouse metabolic enzymes like Cyp3a in conventional chimeric mice with humanized liver, such as PXB-mice [urokinase plasminogen activator/severe combined immunodeficiency (uPA/SCID) mice repopulated with over 70% human hepatocytes] to improve the prediction of drug metabolism and pharmacokinetics in humans. After human hepatocytes were transplanted into Cyp3a KO/uPA/SCID host mice, human albumin levels logarithmically increased until approximately 60 days after transplantation, findings similar to those in PXB-mice. Quantitative real-time–polymerase chain reaction analyses showed that hepatic human P450s, UGTs, SULTs, and transporters mRNA expression levels in Cyp3a KO chimeric mice were also similar to those in PXB-mice and confirmed the absence of Cyp3a11 mRNA expression in mouse liver and intestine. Findings for midazolam and triazolam metabolic activities in liver microsomes were comparable between Cyp3a KO chimeric mice and PXB-mice. In contrast, these activities in the intestine of Cyp3a KO chimeric mice were attenuated compared with PXB-mice. Owing to the knockout of murine Cyp3a, hepatic Cyp2b10 and 2c55 mRNA levels in Cyp3a KO/uPA/SCID mice (without hepatocyte transplants) were 8.4- and 61-fold upregulated compared with PXB-mice, respectively. However, human hepatocyte transplantation successfully restored Cyp2b10 level nearly fully and Cyp2c55 level partly (still 13-fold upregulated) compared with those in PXB-mice. Intestinal Cyp2b10 and 2c55 were also repressed by human hepatocyte transplantation in Cyp3a KO chimeric mice.
Introduction
Liver is the principal site for drug metabolism by virtue of its high expression of enzymes. Among these enzymes, cytochrome P450 enzymes (P450s) play central roles in the oxidative metabolism of a vast range of xenobiotic compounds, including medical drugs. Because of the significant species differences in many hepatic enzymes, the metabolism of drug candidates is normally evaluated in vitro using human liver microsomes or isolated hepatocytes (Brandon et al., 2003; Gómez-Lechón et al., 2003). Although significant advancements in human biologic materials have been made, in vitro test systems have only limited use in predicting human drug metabolism in vivo, particularly for sequential metabolism, owing to the multiple drug-metabolizing enzymes involved (Anderson et al., 2009; Dalvie et al., 2009). Thorough evaluation of the safety and drug-drug interaction of these metabolites will require more accurate methods of predicting human metabolites in the early stages of drug development.
Several mouse models have been developed recently as in vivo test systems that closely resemble clinical conditions. These models insert and express human drug metabolism-related genes such as CYP1A1/2, CYP2D6, CYP2E1, and CYP3A4 (Muruganandan and Sinal, 2008) or the CYP3A cluster (Kazuki et al., 2013). While this transgenic approach has proven useful to a degree, a number of factors may induce inappropriate expression of the transgene, and even if the transgene is expressed appropriately, the remaining cellular components—such as inhibitors and activators and the corresponding cofactors—are all derived from mice, resulting in failure of P450s to interact efficiently or effectively with human genes/gene products.
An alternate approach involves producing chimeric mice with humanized liver by transplanting human hepatocytes into liver-injured immunodeficient mice that can accept xenografted human hepatocytes in their liver. This process takes a few months to produce chimeric mice highly repopulated with human hepatocytes. Several different methods of inducing liver injury and different immunodeficiencies have been investigated (Peltz, 2013), and chimeric mice with humanized liver generated using urokinase-type plasminogen activator/severe combined immunodeficiency (uPA/SCID) mice repopulated with human hepatocytes (PXB-mice; PhoenixBio Co., Ltd., Hiroshima, Japan) have been reported (Tateno et al., 2004). These mice are repopulated with over 70% of human hepatocytes. Expression levels and activities of P450s and non-P450 enzymes, such as uridine 5′-diphospho-glucuronosyl transferase (UGT) and sulfotransferase (SULT), in the liver of PXB-mice are similar to those in humans (Katoh et al., 2004, 2005; Nishimura et al., 2005; Katoh and Yokoi, 2007; Kitamura et al., 2008), and human-specific metabolites are also formed in PXB-mice (Inoue et al., 2009; Kamimura et al., 2010; Yamazaki et al., 2010; De Serres et al., 2011).
However, the utility of chimeric mice with humanized liver as experimental models depends on the extent of replacement of host hepatocytes with transplanted human hepatocytes, as the presence of residual mouse hepatocytes hinders determination of the phenotypes of human hepatocytes. Even in chimeric mice with high hepatic replacement ratios, metabolism profiles are often similar to those in the control mice depending on the compounds, a phenomenon that occurs when examining compounds with metabolic rates in mice higher than in humans, thereby suggesting some degree of metabolism by the remaining mouse hepatocytes in the liver or host animal small intestine, which acts as an extrahepatic metabolism organ (Kamimura et al., 2010). Given the above, mice with their hepatocytes completely replaced by human hepatocytes would be ideal for hepatic metabolism studies, but no group has yet been able to successfully rear these mice.
In humans, CYP3A contributes to the oxidative/reductive metabolism of 50% of drugs used in the clinic and is the most abundant P450 isozyme in human liver and small intestine (Guengerich, 1999; Hall et al., 1999; Dresser et al., 2000). In mice, eight Cyp3a isozymes (Cyp3a11, 3a13, 3a16, 3a25, 3a41, 3a44, 3a57, and 3a59) are known, with Cyp3a13 being mainly expressed in small intestine and Cyp3a11, 3a25, and 3a41 in liver (Martignoni et al., 2006). The tissue distribution and substrate specificities of these CYP3A and Cyp3a enzymes show very extensive overlap. Therefore, as one approach to overcoming the issues of concomitant mouse metabolic activities, we have developed murine Cyp3a knockout (KO) chimeric mice with humanized liver by crossing murine Cyp3a cluster-KO mice with uPA gene–introduced SCID mice. Human hepatocytes were transplanted into the resulting liver-injured Cyp3a KO immunodeficient mice to prepare Cyp3a KO chimeric mice.
Here, to investigate human-P450 and murine-P450 mRNA expressions as well as midazolam and triazolam metabolic activities in liver and intestine, and human UGT, SULT, and transporter mRNA expressions in liver, we describe initial characterizations of Cyp3a KO chimeric mice and compare our findings with those in PXB-mice.
Materials and Methods
Chemicals.
Midazolam, alprazolam, and triazolam were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). 1′- and 4-hydroxymidazolam were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). 1′-hydroxytriazolam and 1′ -hydroxytriazolam-d4 were purchased from Cerilliant Corp. (Round Rock, TX). Gefitinib was purchased from Selleckchem (Houston, TX). All other reagents and solvents used were commercially available and of guaranteed purity.
Generation of Cyp3a–/–/uPA+/+/SCID+/+ Mice.
Cyp3a13–/– lines and Cyp3a57-59–/– lines (lacking the rest of seven Cyp3a gene cluster) were generated in a previous study, and the two lines were mated to generate Cyp3a–/– mice lacking the mouse Cyp3a gene cluster (Kazuki et al., 2013). The generation of uPA+/+/SCID+/+ mice has been described previously (Tateno et al., 2004). Sperm of Cyp3a–/– mice and the unfertilized ovum of uPA+/+/SCID+/+ mice were combined and transferred into foster mothers for artificial insemination. Litters with Cyp3a–/wt/uPA+/wt/SCID+/wt were backcrossed with uPA+/+/SCID+/+ mice via natural mating to obtain litters with Cyp3a–/wt/uPA+/+/SCID+/+. Continuous backcrossing with uPA+/+/SCID+/+ mice was conducted twice via natural mating to obtain Cyp3a–/wt/uPA+/+/SCID+/+ mice. Subsequently, Cyp3a–/wt/uPA+/+/SCID+/+ mice were mated together to obtain Cyp3a–/–/uPA+/+/SCID+/+ mice (designated Cyp3a KO/uPA/SCID mice). Genotype analyses of Cyp3a–/– mice (designated Cyp3a KO mice) and of uPA+/+/SCID+/+ mice (designated uPA/SCID mice) have been described previously (Tateno et al., 2004; Kazuki et al., 2013). The genotypes of Cyp3a and uPA were analyzed by the genomic polymerase chain reaction (PCR) method and the genotypes of SCID by the PCR–restriction fragment length polymorphism method.
Generation of Cyp3a KO Chimeric Mice with Humanized Liver.
Cyp3a KO chimeric mice transplanted with human hepatocytes were prepared by PhoenixBio Co., Ltd. (Hiroshima, Japan). Human hepatocytes of two donors (5-year-old African-American boy, Lot No. BD85, and 2-year-old Hispanic girl, Lot No. BD195) were obtained from Corning Life Sciences K.K. (Tokyo, Japan). The thawing and transplantation of human hepatocytes into CYP3a KO/uPA/SCID mice has been described previously in the generation of PXB-mice (Tateno et al., 2004). Briefly, these mice at 2–4 weeks of age were anesthetized with ether and injected with 2.5–5.0 × 105 viable hepatocytes through a small left-flank incision into the inferior splenic pole.
Generation of PXB-Mice.
PXB-mice transplanted with human hepatocytes of a donor (5-year-old African-American boy, Lot No. BD85) were prepared by PhoenixBio Co., Ltd., as described previously (Tateno et al., 2004).
Measurement of Human Albumin.
Blood samples (2 μl) were collected periodically from the tail vein, and the human albumin (hAlb) level was measured using latex agglutination immunonephelometry (LX Reagent “Eiken” Alb II; Eiken Chemical Co., Ltd., Tokyo, Japan) to predict the replacement index (RI) of human hepatocytes in mouse liver (Tateno et al., 2004).
Immunohistochemistry.
Cryosections prepared from the liver and intestine (5-μm thick) were incubated with anti-human cytokeratin 8 and 18 (CK8/18) mouse monoclonal antibodies (POG-10502; PROGEN Biotechnik GmbH, Heidelberg, Germany) for the liver, or anti-mouse Cyp3a goat polyclonal antibodies (sc-30621; Santa Cruz Biotechnology, Inc., Dallas, TX) for the liver and intestine. The primary antibodies were visualized with Alexa 488- or 594-conjugated donkey anti-mouse-IgG or goat anti-rat IgG (Invitrogen, Carlsbad, CA) as secondary antibodies. The sections prepared from the liver and intestine were stained with Hoechst 33342 for nuclear staining.
Measurements of the Replacement Index.
Measurements of the RI of human hepatocytes to total human and mouse hepatocytes have been described previously (Tateno et al., 2004). Briefly, RI was determined immunohistochemically using anti-human CK8/18 antibodies to calculate the ratio of area occupied by anti-human CK8/18-positive hepatocytes to the entire area examined on immunohistochemical sections.
RNA Extraction and cDNA Synthesis.
Total RNA was extracted from the liver or intestine using the RNeasy minikit (Qiagen, Hilden, Germany), and cDNA was generated from a random hexamer using the high-capacity cDNA reverse transcription kit (Life Technologies, Grand Island, NY).
Quantitative Real-Time–Polymerase Chain Reaction.
Quantitative real-time (qRT)-PCR was conducted using the ABI Prism 7900HT sequence detection system with TaqMan gene expression assays (Life Technologies). The primer and probe sets used were Mm00487224_m1 for Cyp1a2, Mm00456591_m1 for Cyp2b10, Mm00725580_s1 for Cyp2c29, Mm00833845_m1 for Cyp2c37, Mm00472168_m1 for Cyp2c55, Mm00731567_m1 for Cyp3a11, 4652341E for β-Actin, Hs00167927_m1 for CYP1A2, Hs03044634_m1 for CYP2B6, Hs00258314_m1 for CYP2C8, Hs00426397_m1 for CYP2C9, Hs00426380_m1 for CYP2C19, Hs00164385_m1 for CYP2D6, Hs00430021_m1 for CYP3A4, Hs00426592_m1 for UGT2B7, Hs00419411_m1 for SULT1A1, Hs00960941_m1 for SULT1E1, Hs00234219_m1 for SULT2A1, Hs00104491_m1 for MDR1, and 4326315E for β-ACTIN. Other primers and probes used have been described previously for UGT1A1 (Katoh et al., 2005) and UGT1A9 and MRP2 (Nishimura et al., 2002). mRNA levels were normalized against those of β-ACTIN (human) or β-Actin (mouse). The primers for human and murine mRNA were confirmed not to crossreact.
Preparation of Liver and Intestinal Microsomes.
Liver microsomes were prepared as described previously (Sugihara et al., 2001). Briefly, liver samples were homogenized in ice-cold 50 mM Tris-HCl buffer (pH 7.4) and centrifuged at 9,000g for 20 minutes at 4°C. The resulting supernatant was then centrifuged at 105,000g for 60 minutes at 4°C. The microsomal pellet was washed and resuspended in 50 mM Tris-HCl buffer (pH 7.4) and then centrifuged at 105,000g for 60 minutes at 4°C. The final resulting pellet was then suspended in a small amount of 250 mM sucrose and stored at −80°C until analyses. Intestinal microsomes were prepared as described previously (Bonkovsky et al., 1985) with some modifications. The inner space of small intestine samples was washed with ice-cold solution A (pH 7.4, 1.5 mM KCl, 96 mM NaCl, 27 mM sodium citrate, 8 mM KH2PO4, 5.6 mM Na2HPO4 • 12H2O, 230 μM PMSF) and then cut into three parts. These parts were placed in ice-cold solution B (pH 7.4, phosphate-buffered saline supplemented with 1.5 mM EDTA, 0.5 mM DDT, 3 IU/ml heparin, and 230 μM PMSF) and cut longitudinally. Subsequently, the mucosal cells were scraped off the cut samples with a spatula and centrifuged at 2,000g for 5 minutes at 4°C. The centrifuged mucosal cells were added to ice-cold solution C (pH 7.4, 5 mM histidine, 0.25 M sucrose, and 0.5 M EDTA), centrifuged at 2,000g for 10 minutes at 4°C, and then homogenized. The homogenates were centrifuged at 12,000g for 20 minutes at 4°C, and the supernatant was centrifuged at 104,000g for 60 minutes at 4°C. The pellet was mixed with 150 mM Tris and 10 mM EDTA and then centrifuged at 104,000g for 60 minutes at 4°C. The pellet was then resuspended in a small amount of 250 mM sucrose and stored at −80°C until analyses. The protein concentration was determined using a Bradford protein assay kit (Bio-Rad, Hercules, CA) with bovine serum albumin as the standard.
Midazolam Metabolism in Liver and Intestinal Microsomes.
The reaction mixture (200 μl) was composed of 100 mM Na-K phosphate buffer (pH 7.4), 0.1 mM EDTA, 50 μM midazolam, and 0.1 mg protein/ml of liver microsomes or 0.5 mg protein/ml of intestinal microsomes. After 5 minutes of preincubation at 37°C, the reaction was initiated by adding 1 mM NADPH to the final concentration and allowed to proceed at 37°C for 5 minutes for liver microsomes and intestinal microsomes. The reaction was terminated by adding formic acid–50% acetonitrile (10:90 v/v) containing alprazolam (internal standard). The mixture was then centrifuged, and the supernatant was injected into the liquid chromatography–tandem mass spectrometry (LC-MS/MS) system.
Triazolam Metabolism in Liver and Intestinal Microsomes.
The incubations were conducted in a total volume of 200 μl containing 100 mM potassium phosphate buffer (pH 7.4), and 0.5 mg protein/ml liver microsomes or in a total volume of 100 μl containing 100 mM potassium phosphate buffer (pH 7.4), and 1 mg protein/ml of intestinal microsomes. For the stimulation of triazolam metabolism, microsomes were preincubated at 37°C for 4 minutes in the absence and presence of gefitinib (12.5 μM final concentration). Subsequently, triazolam (50 μM final concentration) was added and preincubated at 37°C for 1 minute. The reaction was initiated by adding NADPH-regenerating solution and allowed to proceed at 37°C for 20 minutes for liver microsomes and intestinal microsomes. The reaction was terminated by adding acetonitrile, and 1′-hydroxytriazolam-d4 (internal standard) was added to the incubation mixtures. The mixture was then centrifuged, and the supernatant was injected into LC-MS/MS system.
LC-MS/MS Analysis for 1′-Hydroxymidazolam and 4-Hydroxymidazolam.
The concentrations of 1′-hydroxymidazolam and 4-hydroxymidazolam in microsomal samples were measured using a TSQ Quantum Ultra system (Thermo Fisher Scientific Inc., San Jose, CA). The analytes were separated with a Luna C18(2) column (100 × 2.1 mm, 3 μm; Phenomenex, Torrance, CA) at 40°C and a flow rate of 0.3 ml/min. The mobile phase consisted of 0.1% acetic acid–acetonitrile (90:10 v/v) (A) and acetic acid–acetonitrile (0.1:100 v/v) (B). The gradient condition was linearly increased from 5% to 17% B over 0.5 minutes, maintained at 17% B for 3.5 minutes, linearly increased to 80% B over 1.5 minutes, maintained for 2.5 minutes, and then finally kept at 5% B for 4.5 minutes. The mass spectrometry detection was performed using the positive electrospray ionization selected reaction monitoring mode. The ion spray voltage was set at 4500 V, and the transfer capillary temperature was set at 350°C. The mass spectrometer was set to monitor the transitions of the precursors to the product ions as follows: m/z 342.0 to 324.0 for 1′-hydroxymidazolam, m/z 342.0 to 325.0 for 4-hydroxymidazolam, and m/z 309.0 to 205.0 for alprazolam. Data were processed using Xcalibur software version 1.4.1 (Thermo Fisher Scientific Inc., San Jose, CA). The calibration curves were linear over the range of 7.2 to 13314 nM (liver microsomes) and 1.8 to 3328 nM (intestinal microsomes) for 1′-hydroxymidazolam, and 0.5 to 953 nM (liver microsomes), and 1.2 to 238 nM (intestinal microsomes) for 4-hydroxymidazolam.
LC-MS/MS Analysis for 1′-Hydroxytriazolam.
The concentration of 1′-hydroxytriazolam in microsomal samples was measured using a 4000 QTRAP system (AB SCIEX, Framingham, MA). The analyte was separated with an Inertsil ODS-4 (50 × 2.1 mm, 3 μm) (GL Sciences Inc., Tokyo, Japan) at 40°C and a flow rate of 0.2 ml/min. The mobile phase consisted of formic acid/water (1:1000 v/v) (A) and methanol (B) at a ratio of 35:65 (A/B v/v). The mass spectrometry detection was performed using the positive electrospray ionization multiple reaction monitoring mode. The mass spectrometer was set to monitor the transitions of the precursors to the product ions as follows: m/z 359.0 to 330.9 for 1′-hydroxytriazolam, and m/z 365.2 to 337.1 for 1′-hydroxytriazolam-d4. Data were processed using Analyst software version 1.4.2 (AB SCIEX). The calibration curve was linear over the range of 2.8 to 2784 nM (liver microsomes and intestinal microsomes) for 1′-hydroxytriazolam.
Statistical Analysis.
Data in all tables and figures are shown as the means and standard deviation. Statistical analyses were performed by one-way analysis of variance followed by Tukey’s test using PRISM 5.0 software (GraphPad Software Inc., San Diego, CA) for murine P450 mRNA expressions and midazolam and triazolam metabolic activities, and by t test using WinNonlin 6.1 software (Pharsight Corporation, St. Louis, MO) for triazolam metabolic activity in the presence or absence of gefitinib.
Results
Generation of Cyp3a KO Chimeric Mice with Humanized Liver.
Cyp3a KO mice were crossed with uPA/SCID mice to generate Cyp3a KO/uPA/SCID mice. At 2–4 weeks of age, 2.5, 3.0, 3.5, or 5.0 × 105 viable human hepatocytes were transplanted into Cyp3a KO/uPA/SCID mice to develop Cyp3a KO chimeric mice. In total, 80 transplantations (donor: BD85) and 152 transplantations (donor: BD195) for Cyp3a KO chimeric mice and 255 transplantations (donor: BD85) for PXB-mice were performed, and the body weight and hAlb data of 12-week-old mice are shown in Table 1. Changes in blood hAlb levels in Cyp3a KO chimeric mice and PXB-mice (donor: BD85) are shown in Fig. 1. The hAlb levels increased steadily after transplantation of human hepatocytes and reached a steady state at around 9 weeks or more, depending on the number of transplanted hepatocytes (Fig. 1). Engraftment of human hepatocytes was inferior in the Cyp3a KO host mice compared with the uPA/SCID host mice because hAlb levels of Cyp3a KO chimeric mice were lower than those of uPA/SCID mice when the same numbers of human hepatocytes (2.5 × 105) were transplanted into the host mice (Fig. 1). Mean body weights and hAlb levels in males were higher than those in 12-week-old females of both Cyp3a KO and uPA/SCID chimeric mice, except for Cyp3a KO chimeric mice with 3.5 × 105 BD85 donor cells (Table 1).
Production of chimeric mice using two different donors and two different host mice
Data of body weight and hAlb at 12 weeks old are shown as mean ± S.D.

Changes in hAlb levels in blood of Cyp3a KO chimeric mice and PXB-mice.
Correlation of hAlb levels and the RI of PXB-mice (donor: BD85) is shown in Fig. 2. hAlb levels were exponentially correlated with RI (y = 733927e0.0314x, r2 = 0.912), indicating that a level of more than 6.6 mg/ml hAlb represents RI exceeding 70%. hAlb and RI of Cyp3a KO mice were determined and plotted on the correlation curve between hAlb and RI of uPA/SCID mice, resulting an almost identical correlation (Fig. 2). The yield of chimeric mice was calculated as the portion of chimeric mice with RI > 70% (hAlb > 6.6 mg/ml) among mice transplanted with human hepatocytes. The portion was more than 70% when 2.5 × 105 and 5.0 × 105 BD85 donor cells were transplanted into the uPA/SCID and the Cyp3a KO host mice, respectively, and 3.0 × 105 BD195 donor cells were transplanted into the Cyp3a KO host mice (Table 1).

Correlation between hAlb levels and RI of Cyp3a KO chimeric mice and PXB-mice.
For further animal studies of human P450s and murine P450s expressions (in males and females), human UGTs, SULTs, MDR1, MRP2 expressions (in males), midazolam and triazolam metabolic activities (in males), Cyp3a KO chimeric mice, and PXB-mice (donor: BD85) with 6.3–15.7 mg/ml hAlb (68–97% RI) in males and 5.4–13.2 mg/ml hAlb (64–92% RI) in females were used. Detailed information on chimeric mice used is shown in Table 2.
Chimeric mice used for analyses in the present study
Immunohistochemistry.
Immunohistochemistry of liver transplanted with human hepatocytes and that of the intestine from Cyp3a KO chimeric mice and PXB-mice are shown in Fig. 3. Liver sections prepared from the left lateral lobe in Cyp3a KO chimeric mice (at 10–11 weeks after transplantation) and PXB-mice (at 10–11 weeks after transplantation) were stained with anti-human CK8/18 antibodies and anti-mouse Cyp3a antibodies. The areas of anti-human CK8/18-negative mouse hepatocytes correspond to those of anti-mouse Cyp3a-positive mouse hepatocytes in PXB-mice. In contrast, the areas of anti-human CK8/18-negative mouse hepatocytes correspond to those of anti-mouse Cyp3a-negative mouse hepatocytes in Cyp3a KO chimeric mice, suggesting that mouse Cyp3a enzymes are not expressed in Cyp3a KO chimeric mice and that human hepatocytes are replaced in the host liver.
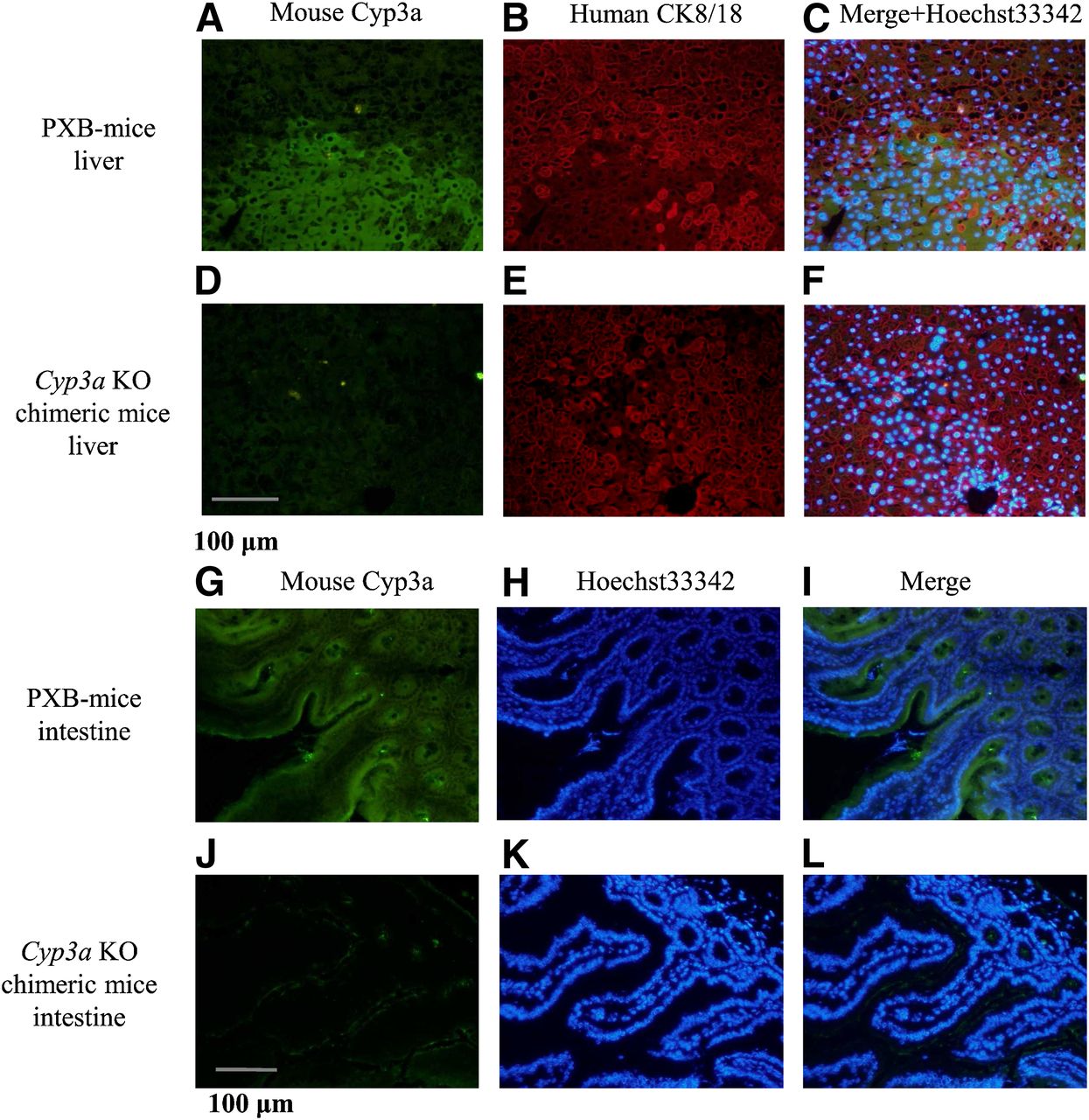
Immunohistochemistry of liver transplanted with human hepatocytes and intestine from Cyp3a KO chimeric mice and PXB-mice. Sections (5-μm thick) of the liver from PXB-mice (A–C) and Cyp3a KO chimeric mice (D–F), and the intestine from PXB-mice (G–I), and Cyp3a KO chimeric mice (J–L) were stained with anti-mouse Cyp3a goat polyclonal antibody for A, D, G, and J in green, anti-human cytokeratin 8 and 18 (CK8/18) mouse monoclonal antibody for B and E in red, and Hoechst 33342 for H and K in blue. In addition, sections A and B or D and E were merged with Hoechst 33342 for C or F, and sections G and H or J and K were merged for I or L, respectively.
Intestinal sections prepared from the same mice were stained with anti-mouse Cyp3a antibodies. Although intestinal section areas in PXB-mice were anti-mouse Cyp3a-positive, those in Cyp3a KO chimeric mice were anti-mouse Cyp3a-negative, suggesting that mouse Cyp3a enzymes are not expressed in the host intestine.
Human and Murine P450s Expression.
Human P450s (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4) and murine P450s (Cyp1a2, 2b10, 2c29, 2c37, 2c55, and 3a11) mRNA expression levels in the liver from PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice are shown in Figs. 4 and 5, respectively. In addition, murine P450s (Cyp2b10, 2c55, and 3a11) mRNA expression levels in the intestine are shown in Fig. 6. Liver and intestine samples from Cyp3a KO chimeric mice and PXB-mice at 10–11 weeks after hepatocyte transplantation and those from Cyp3a KO/uPA/SCID mice without hepatocyte transplantation were used from three male and three female mice. mRNA expression levels were determined via qRT-PCR and normalized using human β-ACTIN for human P450s or murine β-actin for murine P450s. In the liver, human P450s expression levels in Cyp3a KO chimeric mice were comparable (approximately 70–117% in males and 79–122% in females) to those in male PXB-mice.

Human P450 mRNA expression in liver of PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice. Human P450 mRNA expression was determined via qRT-PCR and normalized using the mRNA expression of human β-ACTIN. The primer for human P450s and β-ACTIN used in the present study was confirmed to not crossreact with mouse mRNA. Data are shown as mean + S.D. obtained from three male mice (A–G) and three female mice (H–N). ND, not detected.

Murine P450 mRNA expression in liver of PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice. Murine P450 mRNA expression was determined via qRT-PCR and normalized using the mRNA expression of murine β-actin. The primer for murine P450s and β-actin used in the present study was confirmed not to crossreact with human mRNA. Data are shown as mean + S.D. obtained from three male mice (A–F) and three female mice (G–L). Statistical differences in three mice groups were determined using one-way analysis of variance followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001 versus Cyp3a KO/uPA/SCID mice; ND, not detected.

Murine P450 mRNA expression in the intestine of PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice. Murine P450 mRNA expression was determined via qRT-PCR and normalized using the mRNA expression of murine β-actin. Data are shown as mean + S.D. obtained from three male mice (A–C) and three female mice (D–F). Statistical differences in three mouse groups were determined using one-way analysis of variance followed by Tukey’s test. *P < 0.05 versus Cyp3a KO/uPA/SCID mice. ND, not detected.
The representative isozyme of murine Cyp3a, Cyp3a11 mRNA level was not detected in any liver or intestine samples from Cyp3a KO chimeric mice or Cyp3a KO/uPA/SCID host mice. Hepatic Cyp2b10, 2c29, 2c37, and 2c55 mRNA levels in Cyp3a KO/uPA/SCID mice (without hepatocyte transplant) were 8.4-, 35-, 5.6-, and 61-fold upregulated in males compared with PXB-mice, respectively, owing to the knockout of murine Cyp3a. In Cyp3a KO chimeric mice, hepatic Cyp2b10 and 2c37 mRNA levels were nearly fully restored to those in PXB-mice, but Cyp2c29 and 2c55 levels were 6.1- and 13-fold upregulated in males, respectively, following human hepatocyte transplantation. Intestinal Cyp2b10 and 2c55 mRNA levels in Cyp3a KO/uPA/SCID mice were 2.3- and 51-fold upregulated in males compared with PXB-mice, respectively, owing to the knockout of murine Cyp3a. In Cyp3a KO chimeric mice, intestinal Cyp2b10 and 2c55 mRNA levels were repressed in the same manner as hepatic Cyp2b10 and 2c55 levels by hepatocyte transplantation. Hepatic Cyp1a2, 2b10, and 2c37 mRNA levels did not differ significantly between Cyp3a KO chimeric mice and PXB-mice.
In females, comparable levels of hepatic and intestinal P450 mRNA were detected without any remarkable sex differences.
Midazolam 1′ - and 4-Hydroxylation Activities.
Midazolam 1′- and 4-hydroxylation activities in liver and intestinal microsomes prepared from PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice are shown in Table 3. In liver microsomes, both midazolam 1′- and 4-hydroxylation activities in Cyp3a KO chimeric mice were comparable to those in PXB-mice, although those in Cyp3a KO/uPA/SCID mice (without hepatocyte transplant) were lower owing to the knockout of murine Cyp3a. In intestinal microsomes, both midazolam 1′- and 4-hydroxylation activities in Cyp3a KO chimeric mice were much lower than those in PXB-mice and nearly equal to those in Cyp3a KO/uPA/SCID host mice.
Metabolic activities of 1′- and 4- hydroxymidazolam in liver and intestinal microsomes
Triazolam 1′-Hydroxylation Activity.
Triazolam 1′-hydroxylation activity in liver and intestinal microsomes prepared from PXB-mice, Cyp3a KO chimeric mice, Cyp3a KO/uPA/SCID mice, and uPA/SCID mice as well as humans (liver microsomes only) is shown in Table 4. In addition, triazolam was coincubated with gefitinib to investigate whether triazolam 1′-hydroxylation activity was stimulated in both liver and intestinal microsomes. In liver microsomes, triazolam 1′-hydroxylation activity in Cyp3a KO chimeric mice was comparable to that in PXB-mice and humans, whereas the lower activity in Cyp3a KO/uPA/SCID mice (without hepatocyte transplant) was attributed to the knockout of murine Cyp3a. After coincubation with gefitinib, triazolam 1′-hydroxylation activity was significantly stimulated in PXB-mice and humans, and tended to increase in Cyp3a KO chimeric mice, and significantly inhibited in uPA/SCID mice. In intestinal microsomes, triazolam 1′-hydroxylation activity was not detected in either Cyp3a KO chimeric mice or Cyp3a KO/uPA/SCID host mice.
Metabolic activity of 1′-hydroxytriazolam without or with gefitinib in liver and intestinal microsomes
Human UGTs and SULTs Expression.
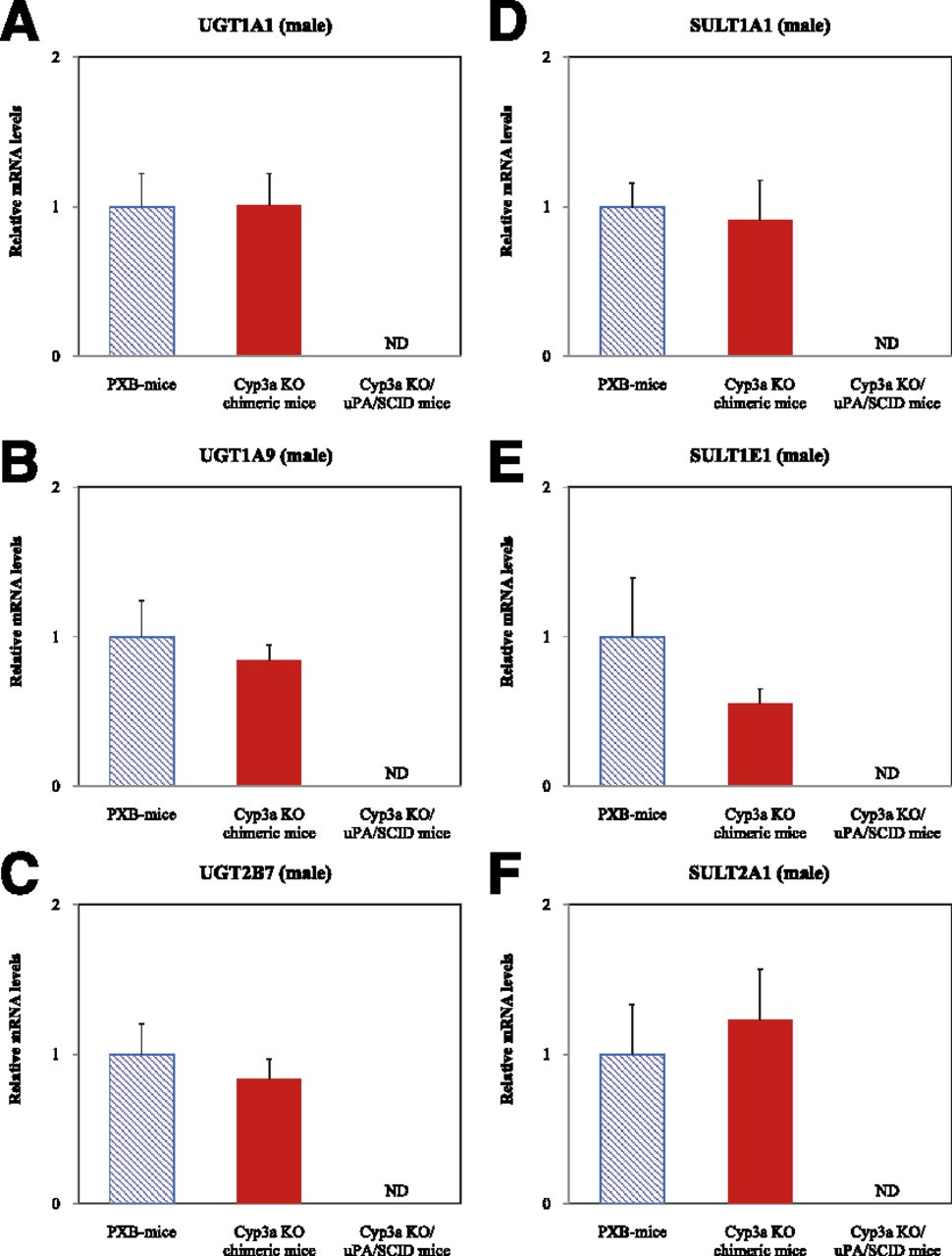
Human UGTs (UGT1A1, 1A9, and 2B7) and SULTs (SULT1A1, 1E1, and 2A1) mRNA expression levels in the liver from PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice are shown in Fig. 7. Hepatic UGT and SULT mRNA levels did not differ significantly between Cyp3a KO chimeric mice and PXB-mice.

Human UGT and SULT mRNA expression in liver of PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice. Human UGT and SULT mRNA expression was determined via qRT-PCR and normalized using the mRNA expression of human β-ACTIN. Primers for human UGTs, SULTs, and β-ACTIN used in the present study were confirmed not to crossreact with mouse mRNA. Data are shown as mean + S.D. obtained from three male mice (A–F). ND, not detected.
Human MDR1 and MRP2 Expression.
Human MDR1 and MRP2 mRNA expression levels in the liver from PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice are shown in Fig. 8. Hepatic MDR1 and MRP2 mRNA levels did not differ significantly between Cyp3a KO chimeric mice and PXB-mice.

Human MDR1 and MRP2 mRNA expression in liver of PXB-mice, Cyp3a KO chimeric mice, and Cyp3a KO/uPA/SCID mice. Human MDR1 and MRP2 mRNA expression was determined via qRT-PCR and normalized using the mRNA expression of human β-ACTIN. Primers for human MDR1, MRP2, and β-ACTIN used in the present study were confirmed not to crossreact with mouse mRNA. Data are shown as mean + S.D. obtained from three male mice (A, B). ND, not detected.
Discussion
We successfully developed Cyp3a KO chimeric mice with humanized liver via transplantation of human hepatocytes into liver-injured Cyp3a KO immunodeficient mice, which were generated through the conventional approach of crossing Cyp3a cluster KO mice with uPA gene–introduced SCID mice. The initial characterization of Cyp3a KO chimeric mice was conducted to investigate human P450s and murine P450 mRNA expression levels, as well as midazolam and triazolam metabolic activities in liver and intestine, and human UGT, SULT, and transporter mRNA expression levels in liver, and then these findings were compared with PXB-mice.
In our initial attempt to generate Cyp3a KO chimeric mice, the replacement of mouse hepatocytes with human hepatocytes after transplantation was much less successful than in PXB-mice (data not shown), probably owing to differences in background genes between Cyp3a KO mice and uPA/SCID mice. Therefore, several continuous backcrosses with uPA/SCID mice were applied to increase the genetic background of the uPA/SCID mice. While engraftment and/or growth of human hepatocytes in the Cyp3a KO host mice were inferior to the PXB-host mice, hAlb levels of Cyp3a KO chimeric mice transplanted with 5.0 × 105 human hepatocytes were similar to those of PXB-mice transplanted with 2.5 × 105 human hepatocytes. In addition, engraftment and/or growth of human hepatocytes were different between BD85- and BD195-transplanted Cyp3a KO chimeric mice. The chimeric mice transplanted with BD195 donor cells showed higher hAlb levels in the blood than those transplanted with BD85 donor cells.
The RI, which was calculated by immunostaining using anti-human CK8/18 antibodies, was correlated with hAlb concentrations in the blood of PXB-mice from a previous study (Tateno et al., 2004) and also those of Cyp3a KO chimeric mice in the present study. Of note, the higher cell number for transplantation resulted in higher hAlb levels in Cyp3a KO chimeric mice.
One of the major limitations inherent to current PXB-mice is the incomplete replacement of mouse hepatocytes with human hepatocytes (Yoshizato et al., 2012). One example of problematic cases is that of chimeric mice administered a drug candidate that is well metabolized by mouse hepatocytes but metabolized only to a limited extent by human hepatocytes, and all its human metabolites are detected as its mouse metabolites (Kamimura et al., 2010). In such a case, the plasma metabolic profile is similar to that of control mice, and no significant increase in the peaks of human metabolites is found. To overcome the metabolic activity caused by residual mouse hepatocytes, we eliminated the contribution of residual host mouse hepatocytes from the currently available PXB-mice by generating Cyp3a KO chimeric mice.
To date, the usefulness of PXB-mice has been recognized in studies for the prediction of human metabolism of drugs (Yoshizato and Tateno, 2009a, b). In the liver of PXB-mice, the human enzymes that play important roles in drug metabolisms include several P450 enzymes, such as CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, and 3A5 and non-P450 enzymes such as UGT, SULT, glutathione S-transferase, N-acetyltransferase, and aldehyde oxidase (Katoh et al., 2004, 2005; Nishimura et al., 2005; Katoh and Yokoi, 2007; Kitamura et al., 2008). In the present study, mRNA levels of selected P450s (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4), UGTs (UGT1A1, 1A9, and 2B7), and SULTs (SULT1A1, 1E1, and 2A1) were measured in the liver to determine whether Cyp3a KO mice are as useful as PXB-mice in drug metabolism and pharmacokinetic research. In addition, as the murine Cyp3a cluster was knocked out, expression levels of several murine P450s (Cyp1a2, 2b10, 2c29, 2c37, 2c55, and 3a11) were measured in the liver and intestine to investigate the impact of the expression of other murine P450s. Given the correlation between hAlb levels and the RI, we concluded that the expression levels of human P450s, UGTs, and SULTs after the transplantation of human hepatocytes into mice did not differ significantly between Cyp3a KO chimeric mice and PXB-mice. Although the expression levels of limited human metabolizing enzymes have been investigated, our results suggest that the Cyp3a KO chimeric mouse is a viable model for drug metabolism and pharmacokinetics research in a manner similar to PXB-mice.
In contrast, significant increases in the expression levels of such murine P450s as Cyp2b10, 2c29, 2c37, and 2c55 in the liver and Cyp2c55 in the intestine were observed in Cyp3a KO/uPA/SCID mice. These findings agree with those of previous reports that found the deletion of the murine Cyp3a cluster to be associated with considerable compensatory changes in other gene families involved in drug metabolism and pharmacokinetics (van Waterschoot et al., 2009b). Interestingly, the upregulated hepatic and intestinal Cyp2b10 expression levels observed in Cyp3a KO/uPA/SCID mice (without human hepatocytes) were restored to near-normal levels in Cyp3a KO chimeric mice that received transplanted human hepatocytes, and remarkable reductions in the expression of hepatic Cyp2c29 and 2c37, and hepatic and intestinal Cyp2c55 from upregulated levels, were also observed, suggesting that transplanted human hepatocytes were able to compensate in part for the loss of murine Cyp3a enzymes not only in the liver but also in the intestine. Although further investigations are needed, the metabolism of dietary phytochemicals of P450 inducers and/or the synthesis or biotransformation of endogenous substances, such as bile acids, might be involved in this compensatory mechanism because these can directly or indirectly regulate P450 expressions (Zhu et al., 2014).
It has been reported that the biotransformation of midazolam is comparable in both humans and mice, yielding 1′-hydroxymidazolam as the major metabolite and 4-hydroxymidazolam as a minor metabolite, and that 1′-hydroxymidazolam formation in mice is not only dependent on murine Cyp3a but also imbued with significant murine Cyp2c components, whereas 4-hydroxymidazolam formation is considered specific to murine Cyp3a (van Waterschoot et al., 2008). Consistent with that report, the present study demonstrated that midazolam 1′-hydroxylation in the liver microsomes of Cyp3a KO/uPA/SCID mice was detectable with partial compensation, probably via induced murine Cyp2c enzymes following murine Cyp3a knockout, and that 4-hydroxylation in the liver microsomes of Cyp3a KO/uPA/SCID mice was reduced. Further, relatively little midazolam metabolism was detected in the intestinal microsomes of either Cyp3a KO chimeric mice or Cyp3a KO/uPA/SCID mice, indicating that the expression level of induced murine Cyp2c enzymes is too low to significantly contribute to the midazolam metabolism in the intestine after the knockout of murine Cyp3a. Midazolam 1′- and 4-hydroxylations in Cyp3a KO chimeric mice were comparable with findings in liver microsomes of PXB-mice after human hepatocyte transplantation, regardless of murine Cyp3a knockout.
In contrast to midazolam, the structurally related drug triazolam is a more specific substrate for CYP3A4 compared with other murine P450 enzymes (Perloff et al., 2000). Although some residual metabolism of triazolam was still mediated by murine Cyp2c enzymes in Cyp3a KO mice, the compensatory triazolam metabolism was much lower compared with midazolam metabolism (van Waterschoot et al., 2009a). From the results in this study, triazolam was metabolized in liver microsomes of uPA/SCID mice but scarcely metabolized in Cyp3a KO/uPA/SCID mice, suggesting involvement of murine Cyp3a in triazolam metabolism of 1′-hydroxylation. Triazolam 1′-hydroxylation in liver microsomes of Cyp3a KO chimeric mice was more than 10-fold higher than that of Cyp3a KO/uPA/SCID mice. The formation of 1′-hydroxylation triazolam was increased by more than twice with gefitinib in liver microsomes of Cyp3a KO chimeric mice, although findings have no significance between those without and with gefitinib in liver microsomes of Cyp3a KO chimeric mice, probably owing to intervariability within the limited number of the mice tested. As it was reported that the anticancer drug gefitinib significantly stimulated triazolam metabolism in human liver microsomes (van Waterschoot et al., 2009a), these results suggest that the triazolam metabolism in Cyp3a KO chimeric mice is attributable to human liver. Triazolam 1′-hydroxylation in Cyp3a KO chimeric mice was comparable to the finding in liver microsomes of PXB-mice after human hepatocyte transplantation, regardless of murine Cyp3a knockout. In intestinal microsomes, 1′-hydroxytriazolam was scarcely formed in any microsomes of murine Cyp3a knock-out mice.
Given the importance of metabolizing enzymes in the intestine to limit hepatic and systemic exposure to orally administered drugs in humanized animal models, the present findings suggest that Cyp3a KO chimeric mice could prove more useful than PXB-mice, because their lack of significant metabolism in the intestine improves predictability of human metabolites. The present study also demonstrated a similarity in the mRNA expression levels of the transporters human MDR1 and MRP2, which both contribute to the distribution and excretion of drugs in both Cyp3a KO chimeric mice and PXB-mice.
In conclusion, we successfully developed Cyp3a KO chimeric mice with humanized liver expressing human P450s similar to PXB-mice but with liver and intestine not expressing murine Cyp3a, a property which may prove useful in drug metabolism and pharmacokinetics research. Further, the method of generating Cyp3a KO chimeric mice described here may be useful for generating humanized animal models with knockout of other murine P450s, non-P450s, and transporters, although the possibility of unexpected side effects (upregulation or downregulation) after knockout of these genes cannot be excluded; as such, any future attempts at generating new models should be conducted with caution.
Acknowledgments
The authors thank Hiroki Ebine, Satoko Yoshida, Miyoko Ono, and Toshihiro Sasaki in Sekisui Medical Co., Ltd. for triazolam metabolism study using liver and intestinal microsomes.
Authorship Contributions
Participated in research design: Kato, Kawamura, Kazuki, Oshimura, Kamimura, Tateno.
Conducted experiments: Ohbuchi, Hamamura, Ohshita.
Performed data analysis: Kato, Ohbuchi, Hamamura, Ohshita.
Wrote or contributed to the writing of the manuscript: Kato, Ohbuchi, Nakada, Sato, Kawamura, Usui, Kazuki, Oshimura, Kamimura, Tateno.
Footnotes
- Received January 22, 2015.
- Accepted May 15, 2015.
Current affiliations:
↵1 Analysis and Pharmacokinetics Research Laboratories, Drug Discovery Research, Astellas Pharma Inc. 2-1-6, Kashima, Yodogawa-ku, Osaka City, Osaka 532-8514, Japan.
↵2 Department of Biomedical Science, Institute of Regenerative Medicine and Biofunction, Graduate School of Medical Science, and Chromosome Engineering Research Center, Tottori University, 86, Nishi-cho, Yonago City, Tottori 683-8503, Japan.
↵3 ADME and Tox Research Institute, Sekisui Medical Co., Ltd., 13-5, Nihonbashi 3-chome, Chuo-ku, Tokyo 103-0027, Japan.
↵4 PhoenixBio Co., Ltd., 3-4-1, Kagamiyama, Higashi-Hiroshima City, Hiroshima 739-0046, Japan.
↵5 Liver Research Project Center, Hiroshima University, 1-2-3, Kasumi, Minami-ku, Hiroshima City, Hiroshima 734-8551, Japan.
Abbreviations
- hAlb
- human albumin
- KO
- knockout
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MDR1
- multidrug resistance protein 1
- MRP2
- multidrug resistance-associated protein 2
- P450
- cytochrome P450
- PMSF
- phenylmethylsulfonyl fluoride
- PXB-mice
- chimeric mice with humanized liver
- qRT-PCR
- quantitative real-time–polymerase chain reaction
- RI
- replacement index
- SCID
- severe combined immunodeficiency
- SULT
- sulfotransferase
- UGT
- uridine 5′-diphospho-glucuronosyl transferase
- uPA
- urokinase-type plasminogen activator
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}