


Abstract
Classical benzodiazepine drugs are in wide clinical use as anxiolytics, hypnotics, anticonvulsants, and muscle relaxants. They act by enhancing the γ-aminobutyric acidA (GABAA) receptor function in the central nervous system. The pharmacological relevance of the multitude of structurally diverse GABAAreceptor subtypes has only recently been identified. Based on an in vivo point mutation strategy, α1-GABAAreceptors were found to mediate sedation, anterograde amnesia, and part of the seizure protection, whereas α2-GABAAreceptors, but not α3-receptors, mediate anxiolysis. Rational drug targeting to specific receptor subtypes has now become possible. Only restricted neuronal networks will be modulated by the new subtype-selective drugs. Promising new anxiolytics have already been developed. A new pharmacology of the benzodiazepine site is on the horizon.
GABAergic inhibition is one of the most rapidly developing topics in neuropharmacology. New therapeutic opportunities arise due to increasing insights into the molecular architecture and diversity of the components involved in signal transduction such as GABAA receptors, GABAB receptors, and GABA transporters (Fig. 1). GABAA receptors are important drug targets representing the sites of action of benzodiazepines, barbiturates, and neurosteroids. The present article focuses on the pharmacological distinction of GABAA receptor subtypes as a basis for the development of new drugs that target restricted neuronal networks. In particular, new ligands of the benzodiazepine site acting selectively on GABAA receptor subtypes are expected to dissect the pharmacological spectrum of classical benzodiazepines and display a minimum of side effects. For further information on GABAA receptor subtypes other recent reviews may be consulted (Barnard et al., 1998; Möhler et al., 2000; Olsen and Homanics, 2000; Whiting et al., 2000; Möhler, 2001).

Scheme of a GABAergic synapse depicting the major elements of signal transduction. GABAA receptors are indirectly linked to the synaptic anchoring protein gephyrin.
Synaptic Action of Benzodiazepines
At synapses, GABAA receptors are activated by a brief nonequilibrium exposure to high concentrations of GABA. Consistent with an increase in the affinity of the receptors for GABA, therapeutically active benzodiazepines prolonged the decay of spontaneous miniature inhibitory postsynaptic currents (mIPSC). Similarly, the amplitude of mIPSC was found to be enhanced by a benzodiazepine agonist in various neuronal systems (Perrais and Ropert, 1999; Hajos et al., 2000), suggesting that the drug-induced increase of the affinity for GABA resulted in the recruitment of more receptors for activation by GABA. However, in other neuronal systems the amplitude of the mIPSC remained unaltered by a benzodiazepine agonist (Mody et al., 1994; Poncer et al., 1996; Hajos et al., 2000), which has been interpreted to indicate that the release of a single quantum of GABA saturates all of the available GABAA receptors in the respective synapses inducing a maximal peak response without further enhancement by the drug. Thus, at synapses that generate mIPSCs, the postsynaptic receptor occupancy by GABA appears to be cell- and synapse-specific, reflecting local differences in the number of receptors or the GABA concentration in the cleft. Accordingly, the influence of benzodiazepine agonists on the amplitude of mIPSCs appears to vary with the operational configuration of the GABAergic synapse (Hajos et al., 2000). In summary, the enhancement of a GABAergic inhibitory response by a benzodiazepine agonist is based on the prolonged decay of the mIPSC and a potential increase of the mIPSC amplitude. Even if the peak mIPSC amplitude is not enhanced per se, the drug-induced prolongation of individual mIPSCs will be reflected in an increased peak amplitude of the compound inhibitory response caused by the summation of several miniature currents (Mody et al., 1994).
GABAA Receptors and Their Multiplicity
Based on the presence of 7 subunit families comprising at least 18 subunits in the central nervous system (α1–6, β1–3, γ1–3, δ, ε, θ, ρ1–3,) the GABAAreceptors display an extraordinary structural heterogeneity. Most GABAA receptor subtypes in vivo are believed to be composed of α-, β-, and γ- subunits. The role of the δ-, ε-, and θ- subunits, which have a very limited expression pattern in the brain, remains to be determined, but it is possible that they substitute for the γ-subunit in α-β-γ combinations. The physiological significance of the structural diversity of GABAA receptors lies in the provision of receptors that differ in their channel kinetics, affinity for GABA, rate of desensitization, and subcellular positioning. In addition, the GABAA receptor subtypes can be distinguished by their pharmacology.
Synaptic and Extrasynaptic GABAA Receptors.
The first electron microscopic studies of GABAAreceptors revealed the ubiquitous presence of extrasynaptic GABAA receptors in the cerebellum, thalamus, and cerebral cortex (Somogyi, 1989). In fact, synaptic GABAA receptors are best seen using postembedding electron microscopy, or by immunofluorescence staining using weakly fixed brain sections (Fritschy et al., 1998a), showing pronounced enrichment in the postsynaptic density of GABAergic synapses compared with extrasynaptic sites (Nusser et al., 1995). These studies also revealed differential targeting of GABAA receptor subtypes to different types of synapses. For instance, the α2 subunit in hippocampal pyramidal cells is concentrated in synapses on the axon-initial segment (Nusser et al., 1996a; Fritschy et al., 1998a), as well as in synapses formed by cholecystokinin-positive basket cells on the soma (Nyı́ri et al., 2001). The α6 subunit, which represents diazepam-insensitive GABAA receptors in the cerebellum can be found both in excitatory and inhibitory synapses in glomeruli of the granule cell layer, as well as extrasynaptically (Nusser et al., 1996b,1999; Sassoè-Pognetto et al., 2000). Finally, GABAA receptors containing the δ-subunit in the cerebellum are exclusively found at extrasynaptic sites (Nusser et al., 1998). Both extrasynaptic receptor types mediate tonic inhibition of neuronal activity (Brickley et al., 1996, 2001).
Synaptic and extrasynaptic GABAA receptors differ in their kinetic properties in line with their distinct functional roles. For instance, extrasynaptic GABAAreceptors containing the δ-subunit in dentate gyrus and cerebellum are tailor-made for tonic inhibition, due to their high affinity for GABA and slow desensitization kinetics (Brickley et al., 1996; Mody and Nusser, 2000). Marked differences in desensitization kinetics have also been reported for synaptic and extrasynaptic receptors in inferior olivary neurons. GABAA receptors containing the α2-subunit are postsynaptic and characterized by rapid desensitization kinetics, whereas extrasynaptic GABAA receptor containing the α3-subunit desensitize very slowly (Devor et al., 2001). Interestingly, such differences are not evident in recombinant expression systems, suggesting the contribution of additional factors regulating GABAA receptor function at synaptic and extrasynaptic sites. A further identification of the molecular composition of GABAergic postsynaptic densities is expected to shed light on the functional regulation of synaptic GABAA receptors (Moss and Smart, 2001).
The demonstration that gephyrin, a synaptic clustering protein initially isolated with glycine receptors, is present in a subset of GABAergic synapses in the retina (SassoèPognetto et al., 1995) prompted the analysis of its role in relation to GABAA receptors. Gephyrin was shown to be required, along with the γ2-subunit, for postsynaptic clustering of major GABAA receptor subtypes (Essrich et al., 1998; Kneussel et al., 1999). A recent study demonstrated by immunoelectron microscopy and double-immunofluorescence staining that gephyrin is colocalized with the vast majority of postsynaptic GABAA receptors throughout the central nervous system (Sassoè-Pognetto et al., 2000), indicating that it represents a useful marker of GABAergic (and glycinergic) synapses.
Diazepam-Sensitive GABAA Receptors.
Receptors containing the α1-, α2-, α3-, or α5- subunits in combination with any of the β-subunits and the γ2-subunit are most prevalent in the brain (Fig. 2). These receptors are sensitive to benzodiazepine modulation. The major receptor subtype is assembled from the subunits α1β2γ2, with only a few brain regions lacking this receptor (granule cell layer of the olfactory bulb, reticular nucleus of the thalamus, spinal cord motoneurons) (Fritschy and Möhler, 1995; Pirker et al., 2000) (Table 1).
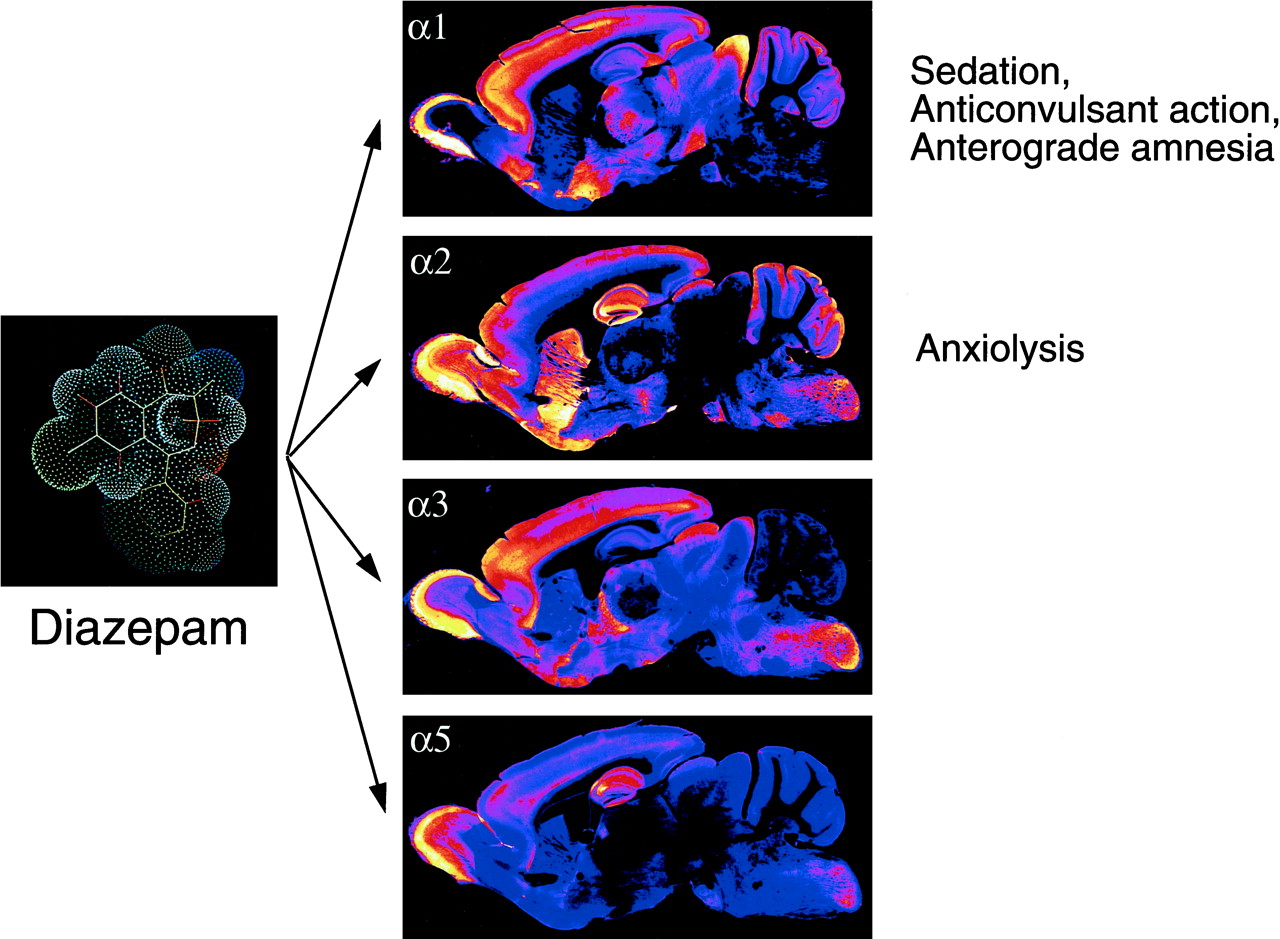
The four classes of diazepam-sensitive GABAA receptors are distinguished by the type of α-subunit (α1, α2, α3, α5) and their largely distinct neuronal localizations as demonstrated immunohistochemically in mouse brain sections. The major known pharmacological actions mediated via the respective receptor subtypes are indicated.
GABAA Receptor Subtypes
Receptors containing the α2- or α3- subunit are considerably less abundant and are highly expressed in brain areas where the α1-subunit is absent or present at low levels (Table 1). The α2- and α3-subunits are frequently coexpressed with the β3- and γ2-subunits, which is particularly evident in hippocampal pyramidal neurons (α2β3γ2) and in cholinergic neurons of the basal forebrain (α3β3γ2). The ligand-binding profile of these receptors differs from that of α1β2γ2 by having a considerably lower displacing potency for ligands such as 3-carboxymethoxy-β-carboline, CL 218,872, and zolpidem (Table1).
Receptors containing the α5-subunit are of minor abundance in the whole brain (Table 1) but are expressed to a significant extent in the hippocampus, where they comprise 15 to 20% of the diazepam-sensitive GABAA receptor population, predominantly coassembled with the β3-and γ2-subunits. The α5-receptors are differentiated from α1β2γ2, α2β3γ2, and α3β3γ2receptors by a lower affinity to CL 218,872 and near insensitivity to zolpidem (Table 1).
The γ1- and γ3-subunits characterize a small population of receptors that contain various types of α- and β-subunits. Due to their reduced affinity for the classical benzodiazepines, they do not appear to contribute to any great extent to their pharmacology in vivo (Möhler et al., 2000;Whiting et al., 2000; Möhler, 2001).
Diazepam-Insensitive GABAA Receptors.
GABAA receptors that do not respond to clinically used ligands, such as diazepam, flunitrazepam, clonazepam, and zolpidem, are of low abundance in the brain and are largely characterized by the α4- and α6-subunits (Table 1). Receptors containing the α4-subunit are generally expressed at very low abundance but more prominently in thalamus and dentate gyrus (Pirker et al., 2000); those containing the α6-subunit are restricted to the granule cell layer of the cerebellum (about 30% of all GABAA receptors in the cerebellum). Both receptor populations are structurally heterogeneous, and the majority of the α6-containing receptors are of the α6β2γ2combination (Table 1). The benzodiazepine site profile of α4- and α6-receptors is characterized by a low affinity for flumazenil and bretazenil and a switch in the efficacy of Ro 15-4513 (ethyl 8-acido-5,6-dihydro-5-methyl-6-oxo-4H-imidazol[1,5-α] [1,4]benzodiazepine-3-carboxylate) from an inverse agonist to an agonist. The δ-subunit is frequently coassembled with the α4- or the α6-subunit in benzodiazepine insensitive receptors (Möhler et al., 2000;Whiting et al., 2000; Möhler, 2001).
In the retina, homomeric receptors consisting of the ρ-subunit represent a particular class of GABA-gated chloride channels. Their GABA site is insensitive to bicuculline and baclofen, and they are not modulated by barbiturates or benzodiazepines (Table 1). Due to these distinctive features the receptors are frequently termed GABAc receptors (Bormann, 2000), although they can also be considered as a homomeric class of GABAA receptors (Barnard et al., 1998).
Distinct Benzodiazepine Sites Per Receptor
The benzodiazepine site is thought to be located at the interface of the respective α-subunit (α1, α2, α3, α5) and the γ2-subunit. About 25% of α1-receptors in rat brain contain a second type of α-subunit, i.e., the α1-subunit is coassembled with the α3-subunit. If there is no preference as to which subunit holds the position adjacent to the γ2-subunit, a mixed pharmacology for the benzodiazepine site would be expected. Receptors with α1 and α3 subunits displayed a ligand-binding profile characteristic of both α3 and α1 subunits with the latter being predominant (Araujo et al., 1996). Therefore, each α subunit appears to contribute its own binding characteristics. For α1α6βγ2receptors, which represent about 40% of all α6receptors (Pollard et al., 1995; Khan et al., 1996; Jechlinger et al., 1998), conflicting results were reported with either each α-subunit contributing its typical pharmacology (Khan et al., 1996) or a single pharmacology typical for α6 (Pollard et al., 1995). Similarly, in radioligand binding assays of hippocampal α5 receptors, only the α5 pharmacology was apparent (Araujo et al., 1999), although the receptors contain an additional α1-, α2-, or α3-subunit (Sur et al., 1998; Araujo et al., 1999). Thus, the impact of distinct α-subunits on the receptor pharmacology remains to be defined.
Recently, a novel, low affinity benzodiazepine site was identified on recombinant GABAA receptors (α1β2γ2) (Walters et al., 2000). It displayed micromolar affinity for diazepam and was insensitive to flumazenil. This site is not of therapeutic relevance since practically all clinically relevant effects of benzodiazepine drugs can be blocked by flumazenil. In addition, the presence of the γ2-subunit was not a prerequisite for the low affinity site since it is also present on α1β1-receptors (Walters et al., 2000). At present it cannot be excluded that the low-affinity site represents the so-called peripheral benzodiazepine site, which is known to occur also on GABAA receptors (Haefely, 1994).
Pharmacology of GABAA Receptor Subtypes in Vivo
In the search for benzodiazepine site ligands with higher therapeutic selectivity and a reduced side effect profile, GABAA receptor subtypes have long been considered to be promising targets. However, it was only recently that the pharmacological relevance of GABAA receptor subtypes was identified based on a gene knock-in strategy (Rudolph et al., 1999; Löw et al., 2000). The benzodiazepine site was rendered diazepam-insensitive by a point mutation in the respective α subunits by replacing a histidine residue with an arginine residue [α1(H101R), α2(H101R), α3(H126R), and α5(H105R)] (Wieland et al., 1992; Benson et al., 1998). Mouse lines were generated in which the α1-, α2-, or α3-receptor subtypes were diazepam-insensitive. In these mice certain benzodiazepine effects were expected to be blunted, which was attributed to the respective point-mutated receptor. This strategy permitted the allocation of the benzodiazepine drug actions to identified GABAA receptor subtypes. In addition, it implicated the neuronal networks expressing the particular receptor in mediating the corresponding drug actions.
The α1-, α2-, and α3-point-mutated receptors displayed a level of expression and a regional and cellular distribution that was indistinguishable from those in wild-type mice. In addition, the gating of the point-mutated receptors by GABA remained unaltered. Furthermore, the neomycin resistance marker introduced by the replacement vector was eliminated from the genome of all mouse lines generated by cre-loxP-mediated recombination. The pharmacological analysis of the point-mutated mice was therefore free of any potential interference, which may have resulted from the presence of the neomycin resistance marker. Thus, a deficit in the behavioral response to diazepam in the point-mutated mouse lines is attributed to the pharmacological role of the respective receptor subtype (Rudolph et al., 1999; Löw et al., 2000).
Receptors Mediating Sedation.
Sedation is a major property of many benzodiazepine site ligands and has now been shown to be mediated via α1-receptors. Among α1-, α2-, and α3-point-mutated mice only the α1(H101R) mutants were resistant to the depression of motor activity by diazepam and zolpidem (Rudolph et al., 1999; Crestani et al., 2000a; Löw et al., 2000). This effect was specific for ligands of the benzodiazepine site since pentobarbital or a neurosteroid remained as effective in α1(H101R) mice as in wild-type mice in reducing motor activity. An α1(H101R) mouse line was also generated by McKernan et al.(2000). When measured under novelty (“stress”) conditions, diazepam increased locomotion compared with wild-type (McKernan et al., 2000). This effect is apparently dependent on the test procedure and can be induced also in the α1(H101R) mice generated by Rudolph et al. (1999) under comparable test conditions (Crestani et al., 2000b).
Receptors Mediating Amnesia.
Anterograde amnesia is a classical side effect of benzodiazepine drugs. The memory-impairing effect of diazepam, analyzed in a step-through passive avoidance paradigm, was strongly reduced in the α1(H101R) mice compared with wild-type mice as shown by the increased latency for reentering the dark compartment 24 h after training (Rudolph et al., 1999). This effect was not due to a potential nonspecific impairment since the ability of a muscarinic antagonist to induce amnesia was retained in the α1(H101R) mice. These results demonstrate that the diazepam-induced anterograde amnesia is mediated by α1-receptors.
Receptors Providing Protection Against Seizures.
The anticonvulsant activity of diazepam, assessed by its protection against pentylenetetrazole-induced tonic convulsions, was reduced in α1(H101R) mice compared with wild-type mice (Rudolph et al., 1999). The anticonvulsant effect of diazepam, which remained in the α1(H101R) mice, was due to GABAA receptors other than α1, since it was antagonized by flumazenil. Sodium phenobarbital remained fully effective as an anticonvulsant in α1(H101R) mice with a dose response intensity similar to that of wild-type mice. These results show that the anticonvulsant activity of benzodiazepines is partially but not fully mediated by α1-receptors. The anticonvulsant action of zolpidem is exclusively mediated by α1-receptors, since its anticonvulsant action is completely absent in α1(H101R) mice (Crestani et al., 2000a).
Receptors for Anxiolysis.
New strategies for the development of daytime anxiolytics that are devoid of drowsiness are of high priority. The particular receptor subtype that mediates the anxiolytic activity of benzodiazepine drugs was recently identified (Löw et al., 2000). When the reactivity to naturally aversive stimuli is measured in wild-type mice, diazepam increases the time spent in the lit area of the light/dark choice test and on the open arms of the elevated plus maze, respectively. In contrast, the α2-point-mutated mice were resistant to the effect of diazepam in these test paradigms (Löw et al., 2000). This lack of response was specific for ligands of the benzodiazepine site since α2(H101R) mice retained the ability to display an anxiolytic-like response to sodium phenobarbital. Thus, the anxiolytic-like action of diazepam is selectively mediated by the enhancement of GABAergic transmission in a population of neurons expressing the α2-GABAAreceptors. Thus, the α2-GABAA receptors are highly specific targets for the development of future selective anxiolytic drugs. They represent only about 15% of all diazepam-sensitive GABAA receptors. Selective ligands are therefore expected to be devoid of the major side effects that afflict the classical benzodiazepine anxiolytics.
It had previously been assumed that the anxiolytic action of diazepam is based on the dampening of the reticular activating system. It is mainly represented by noradrenergic and serotonergic neurons of the brain stem, which express exclusively α3-receptors. The analysis of the α3-point-mutated mice [α3(H126R)] indicated that the anxiolytic effect of benzodiazepine drugs, measured as described above, is not mediated by α3-receptors (Löw et al., 2000). The reticular activating system therefore does not appear to be a major contributor to anxiolysis. In contrast, the α2-GABAA receptors are highly expressed in cells in the cerebral cortex and the hippocampus, including their pyramidal cells, which display particularly high densities of α2-GABAAreceptors on their axon initial segment (Nusser et al., 1996a; Fritschy et al., 1998a). Thus, controlling the output of these principal neurons may contribute to anxiolysis.
Receptors Mediating Myorelaxation.
The degree of muscle tone can be assessed in the horizontal wire test, in which the ability of the animals to grasp and hang onto a wire is measured. The muscle relaxant effect of diazepam is largely mediated by α2-GABAA receptors, as shown by the failure of diazepam to induce changes in muscle tone in the α2-mutated mouse line (Crestani et al., 2001). It was only at high doses that α3-receptors were also implicated. Besides the limbic system (see above), α2-receptors are highly specifically expressed in the spinal cord, notably in the superficial layer of the dorsal horn and in motor neurons (Bohlhalter et al., 1996) with the latter being most clearly implicated in muscle relaxation. It is important to note that the muscle relaxant effect requires higher doses of diazepam than its anxiolytic activity. This is attributed to a higher receptor occupancy required for muscle relaxation.
Novel Subtype-Selective Benzodiazepine Site Ligands
Among the clinically used ligands of the benzodiazepine site only the hypnotic zolpidem displays a pronounced preferential subtype selectivity (Langer et al., 1992) (Table 1). Additional benzodiazepine site ligands with subtype selectivity are under experimental or clinical investigation (Hood et al., 2000) and include the following agents.
L-838,417.
The benzodiazepine site ligand L-838,417 interacts with comparable affinity with α1-, α2-, and α3-receptors and with only 3-fold lower affinity with α5-receptors. However, it displays a dramatic subtype-selective efficacy. L-838,417 fails to modulate the GABA response at α1-receptors but enhances the GABA response at α2-, α3-, and α5-receptors (McKernan et al., 2000). In line with a lack of α1-receptor activation, L-838,417 showed a high potency in anxiolytic tests (elevated plus maze and fear-potentiated startle) and in anticonvulsant tests (pentylenetetrazole, audiogenic seizures). However, even at doses that occupied 95% of benzodiazepine sites, L-838,417 failed to impair motor performance (rotarod test, chain-pulling test) (McKernan et al., 2000). These findings are a major step forward. Ligands with subtype-selective efficacy, which distinguish α2-, α3-, and α5,-receptors from α1-receptors, provide a new way to develop selective anxiolytics without a sedative component. Further improvement may be achieved by focusing the ligand affinity or efficacy more specifically on α2-receptors (see above).
SL65.1498.
The pyrido-indole-4-carboxamide derivative SL65.1498 (6-fluoro-9-methyl-2phenyl-4-(pyrrolidin-1-yl-carbonyl)-2,9-dihydro-1H-pyrido[3,4-b]indol-1-one) shows higher affinity for α1-, α2-, and α3-GABAA receptors compared with α5-receptors. In addition, it acts as full agonist at α2- and α3-receptors but as a partial agonist at α1-GABAA receptors. In line with its selectivity for the activation of α2- and α3-receptors, the compound showed potent anxiolytic action in animal models (punished lever pressing, punished drinking, elevated plus maze, light/dark test) but did not impair motor coordination (e.g., rotarod) or working memory (Morris water maze) (Scatton et al., 2000).
Zaleplon.
Zaleplon (CL 284,846) is a pyrazolopyrimidine developed for the treatment of insomnia (Sanger et al., 1996). At recombinant receptors, zaleplon binds preferentially to α1-receptors (α1β2γ2) and to receptors containing the γ3-subunit but binds 8- to 20-fold less to α2-, α3-, and α5-receptors (Dämgen and Lüddens, 1999). Thus, zaleplon is largely a ligand with preference for α1-receptors, which is in keeping with its preponderant hypnotic activity. The contribution of its interaction with γ3-receptors is unclear since these receptors are of low abundance in the brain.
Conclusions
The genetic dissection of the pharmacological functions of GABAA receptor subtypes has opened up a new strategy in drug development. The heuristic search for novel ligands at the benzodiazepine site will be replaced by the rational targeting of specific receptor subtypes. The ligand selectivity can be achieved either by a preferential ligand affinity, preferential ligand efficacy, or a mixture of both. Acting at small subpopulations of GABAA receptors, these ligands are expected to lack the major side effects of the classical benzodiazepine drugs. The vision of specific anxiolytics with a reduced side-effect profile may become a reality. In addition, therapeutic indications beyond those of the classical benzodiazepine drugs may emerge from subtype-specific drugs.
Abbreviations
- GABA
- γ-aminobutyric acid
- mIPSC
- miniature inhibitory postsynaptic currents
- Received March 5, 2001.
- Accepted July 5, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}