


Abstract
Epilepsy, one of the most common neurologic disorders, is a major public health issue. Despite more than 20 approved antiepileptic drugs (AEDs), about 30% of patients are refractory to treatment. An important characteristic of pharmacoresistant epilepsy is that most patients with refractory epilepsy are resistant to several, if not all, AEDs, even though these drugs act by different mechanisms. This argues against epilepsy-induced alterations in specific drug targets as a major cause of pharmacoresistant epilepsy, but rather points to nonspecific and possibly adaptive mechanisms, such as decreased drug uptake into the brain by intrinsic or acquired over-expression of multidrug transporters in the blood-brain barrier (BBB). There is accumulating evidence demonstrating that multidrug transporters such as P-glycoprotein (PGP) and members of the multidrug resistance-associated protein (MRP) family are over-expressed in capillary endothelial cells and astrocytes in epileptogenic brain tissue surgically resected from patients with medically intractable epilepsy. PGP and MRPs in the BBB are thought to act as an active defense mechanism, restricting the penetration of lipophilic substances into the brain. A large variety of compounds, including many lipophilic drugs, are substrates for either PGP or MRPs or both. It is thus not astonishing that several AEDs, which have been made lipophilic to penetrate into the brain, seem to be substrates for multidrug transporters in the BBB. Over-expression of such transporters in epileptogenic tissue is thus likely to reduce the amount of drug that reaches the epileptic neurons, which would be a likely explanation for pharmacoresistance. PGP and MRPs can be blocked by specific inhibitors, which raises the option to use such inhibitors as adjunctive treatment for medically refractory epilepsy. However, although over-expression of multidrug transporters is a novel and reasonable hypothesis to explain multidrug resistance in epilepsy, further studies are needed to establish this concept. Furthermore, there are certainly other mechanisms of pharmacoresistance that need to be identified.
Pharmacoresistance to Antiepileptic Drugs in Patients with Epilepsy
Epilepsy or the epilepsies are common neurological disorders, affecting approximately 1 to 2% of the population (Browne and Holmes, 2001). Epilepsy is a chronic and often progressive brain disorder, characterized by the periodic and unpredictable occurrence of seizures, which may be generalized, originating simultaneously in both hemispheres of the brain, or partial (focal), originating in one or more parts of one or both hemispheres, most commonly the temporal lobe. Despite considerable progress in understanding the pathogenesis of seizures and epilepsy, for many seizure types and epilepsy syndromes we have little information about their pathophysiological basis (Lothman, 1996; Löscher, 2001). In the absence of a specific etiological understanding, approaches to drug therapy of epilepsy must necessarily be directed at the control of symptoms, i.e., the suppression of seizures. Chronic administration of antiepileptic (anticonvulsant) drugs (AEDs) is the treatment of choice in epilepsy. The selection of an AED is based mainly on its efficacy for specific types of seizures, tolerability, and safety (Browne and Holmes, 2001). The goal of therapy is to keep the patient free of seizures without interfering with normal brain function. In the majority of patients, this goal is reached. However, in about 30% of patients with epilepsy the seizures persist despite the choice of an adequate AED and carefully monitored treatment (Regesta and Tanganelli, 1999). Although the terms “pharmacoresistant” or “medically refractory” lack a precise definition, most clinicians would consider an epilepsy pharmacoresistant that had not been controlled by any of two to three first-line AEDs usually used for a given epilepsy syndrome. The probability of intractability largely depends on the type of seizures and epilepsy, with complex partial seizures such as those occurring in temporal lobe epilepsy having the poorest prognosis of all seizure types in adults (Regesta and Tanganelli, 1999).
Pharmacoresistant epilepsy is a major health problem, associated with increased morbidity and mortality, and accounting for much of the economic burden of epilepsy (Regesta and Tanganelli, 1999). The problem of intractable or difficult-to-treat seizures has not been changed to any significant extent by the recent introduction of various new AEDs, although drug treatment has become more tolerable for a number of patients (Regesta and Tanganelli, 1999; Löscher, 2002). A striking obstacle in developing new strategies for treatment of pharmacoresistant epilepsy is that mechanisms of pharmacoresistance are only poorly understood. Some clinical features are associated with resistance, including early onset of seizures (before 1 year of age), high seizure frequency prior to onset of treatment, a history of febrile seizures, the type of seizures (about 60% of patients with intractable epilepsy suffer from partial seizures) or epilepsy, structural brain lesions, and malformations of cortical development (Regesta and Tanganelli, 1999). However, little research has been undertaken into the basis of these associations.
There are many possible causes of refractory epilepsy; it is likely to be a multifactorial process (Regesta and Tanganelli, 1999). Genetic factors, e.g., polymorphisms, may be important and explain why two patients with the same type of epilepsy or seizures may differ in their response to AEDs. Disease-related factors are certainly important, including the etiology of the seizures, progression of epilepsy under treatment with AEDs, alterations in drug targets, or alterations in drug uptake into the brain. Furthermore, drug-related factors are most likely involved in insufficient seizure control, including loss of anticonvulsant efficacy during treatment, i.e., development of tolerance, or ineffective mechanisms of action of currently available AEDs in patients with medically intractable epilepsy.
An important characteristic of pharmacoresistant epilepsy is that most patients with refractory epilepsy are resistant to most, and often all, AEDs (Regesta and Tanganelli, 1999). As a consequence, patients not controlled on monotherapy with the first AED have a chance of only about 10% or lower to be controlled by other AEDs, even when using AEDs that act by diverse mechanisms. This argues against epilepsy-induced alterations in specific drug targets as a major cause of pharmacoresistant epilepsy, but rather points to nonspecific and possibly adaptive mechanisms, such as decreased drug uptake into the brain by seizure-induced over-expression of multidrug transporters in the blood-brain barrier.
Multidrug Transporters in the Blood-Brain and Blood-Cerebrospinal Fluid (CSF) Barriers
For drugs to enter the brain, they must traverse either the blood-brain barrier (BBB) or the barrier between blood and CSF. Because of these anatomical barriers, entry of drugs into the brain is restricted (Betz et al., 1994; Pardridge, 1999). The restrictive nature of the brain microvessel endothelial cells that form the BBB is due in part to the formation of tight junctions between the cells (see Fig.1), to the lack of transendothelial pathways such as transcellular channels or fenestrations, and to a relative paucity of pinocytotic vesicles (Betz et al., 1994). The functional consequence is that brain capillaries act in a passive manner like continuous phospholipid membranes, largely restricting the penetration of hydrophilic, polar, large, or protein-bound compounds, whereas nonpolar (nonionic), highly lipid-soluble drugs penetrate easily through the BBB by passive diffusion (Fig. 1). Surrounding the capillary endothelial cells of the BBB is a collagen-containing extracellular matrix or basement membrane, which is covered with foot processes from astrocytes (see Fig. 1). The astrocytic investment of blood vessels in the brain has suggested a role in the BBB system, but under normal conditions the astrocytic end-feet provide little resistance to the movement of molecules (Betz et al., 1994). With respect to the blood-CSF barrier (BCB), for a drug to enter the CSF it must pass through the choroid plexus (CP). Because capillary endothelial cells of the CP are fenestrated and lack tight junctions, the permeation barrier within the CP exists at the level of the epithelial cells lining the surface (Betz et al., 1994). These epithelial cells are mainly joined by tight junctions, which restrict entry of water-soluble molecules (Betz et al., 1994; Spector, 2000).

A schematic representation of the BBB and the role of multidrug transporters (mdt) in drug transport through the BBB. A brain capillary with three endothelial cells separated by tight junctions is shown. The capillary endothelium is surrounded by a basement membrane (not illustrated) and a sheath of astrocytic processes (glial end-feet). The functional consequence of these features is that brain capillaries act in a passive manner to restrict penetration of hydrophilic or large substances, but highly lipophilic drugs (like most antiepileptic drugs) penetrate easily through the BBB by simple diffusion (dashed arrows). As an active defense mechanism of the BBB against lipophilic substances, ATP-dependent multidrug transporters, such as PGP or MRP2, which are located in the apical (luminar) cell membrane of capillary endothelial cells, act as outwardly directed active efflux pumps, transferring part of the drug, which has entered the cells by diffusion, back into blood, thus limiting penetration of many lipophilic drugs into brain parenchyma. Furthermore, by lowering drug concentration in the endothelial cells, multidrug transporter proteins may indirectly promote flux from the brain extracellular space into endothelial cells (dashed arrow), followed by extrusion into the blood (Edwards, 2001). In epileptogenic brain tissue, these transporters are over-expressed in capillary endothelial cells and astrocytes around blood vessels, so that now the glial end-feet may contribute to the barrier function as a “second line defense” mechanism (not illustrated).
For many years, the BBB was considered to be an anatomical barrier that absolutely restricts the passage of certain substances into the brain. However, apart from passive diffusion, drugs may also enter and leave the brain by carrier-mediated transport processes (Pardridge, 1999;Spector, 2000). In this respect, the recent finding of multidrug transporters of the ATP-binding cassette superfamily, such as P-glycoprotein (PGP) and multidrug resistance-associated protein (MRP), in the endothelial cells of the BBB (see Fig. 1) is of particular interest, since these outwardly directed active efflux mechanisms appear to act as an active defense mechanism, limiting brain accumulation of many lipophilic drugs (Fromm, 2000; Spector, 2000;Abbott et al., 2002). Furthermore, both PGP and MRP are expressed in CP epithelial cells that form the BCB (Rao et al., 1999).
PGP, a transmembrane glycoprotein active efflux system discovered in 1976, consists of a group of closely related, intrinsic membrane proteins encoded by a small family of genes (Leveille-Webster and Arias, 1995). The PGP isoforms involved in multidrug resistance are encoded by the MDR1 gene in humans and the mdr1aor mdr1b genes in rodents (Leveille-Webster and Arias, 1995). The tissue distribution of these proteins suggests that the two rodent PGP isoforms together perform the same functions as the single human PGP (MDR1) protein. The mdr1-type PGP (also termed PGP-170 because of its molecular weight of 170 kDa) functions as a drug efflux pump with the property of being able to accept a wide range of structurally different hydrophobic substrates, most of which enter cells by passive diffusion (Seelig et al., 2000). PGP, which, like other multidrug resistance proteins, was initially discovered as a membrane transporter producing chemotherapy resistance in a wide range of tumor types, is widely expressed in normal tissues with excretory function such as liver, kidney, and intestine (Fromm, 2000), and is involved in barrier functions such as in the BBB and the blood-testis barrier (Sarkadi et al., 1996). PGP thus is thought to exert a physiological function in normal tissues relating to the excretion and/or protection of tissues from naturally occurring toxins or xenobiotics (Jette et al., 1995). Mice with deletion ofmdr1a or both mdr1a and mdr1b do not show any obvious physiological abnormalities but a marked increase in the brain uptake of various lipophilic drugs, with consequent neurotoxicity (Schinkel et al., 1996, 1997). Most drugs that are good PGP substrates have a molecular weight above 400 Da, which explains that such drugs enter the brain far less efficiently than expected from their lipid solubility (Schinkel, 1999).
The precise location of PGP in the BBB has been a topic of some debate (Schinkel, 1999). The prevailing opinion is that PGP is located in the apical (luminal) cell membrane of capillary endothelial cells as illustrated in Fig. 1 (Schinkel, 1999; Abbott et al., 2002). In contrast, Golden and Pardridge (2000) have proposed that PGP is located primarily in astrocyte foot processes of the BBB. The latter authors proposed that the loss (or inhibition) of this active efflux system at the astrocyte plasma membrane would allow greater drug uptake into the astrocytes. However, previous microdialysis experiments with determination of extracellular brain drug levels after PGP inhibition argue against this assumption because there was a clear increase of drug levels in the extracellular space, which would be in line with the classic model proposed for the function of PGP in the endothelium of the BBB (Burgio et al., 1998; Potschka and Löscher, 2001a,b). Yet, there is some evidence that under pathological conditions, such as epilepsy, PGP in astrocyte foot processes may be involved in BBB function (see below). In the rodent brain, the mdr1a PGP isoform is predominantly expressed in brain microvessel endothelial cells of the BBB, whereas the mdr1b PGP isoform is preferentially expressed in astrocytes (Regina et al., 1998; Decleves et al., 2000). In the normal human brain, PGP is highly expressed in capillary endothelial cells, but cannot be detected by routine immunohistochemistry in brain parenchyma, i.e., astrocytes or neurons (Tishler et al., 1995; Sisodiya et al., 2002). One explanation for the apparent difference in astrocytic PGP expression in rodents and humans could be that PGP in normal human astrocytes is below the detection level of the assays used, because under pathological conditions, such as epilepsy, PGP becomes detectable in human astrocytes (Sisodiya et al., 2002).
The MRP family (with the first member, MRP1, discovered in cancer cells in 1992) currently has seven members (MRP1–7), which act as organic anion transporters, but can also transport neutral organic drugs (Borst et al., 2000). As a consequence, PGP and MRPs have overlapping substrate specificity, so that several drugs are substrates for both transporter families (Borst et al., 2000; Seelig et al., 2000). As PGP, MRPs are located in several normal tissues, including the BBB and BCB (Borst et al., 2000). Some MRPs, like MRP2, are located in apical cell membranes of tissues, which in most membranes is the appropriate position for a protective role, whereas other MRPs, such as MRP1, MRP3, and MRP5, are located basolaterally (Borst et al., 1999). Expression of MRPs in microvessel endothelial cells that form the BBB has been reported only recently (Huai-Yun et al., 1998; Zhang et al., 2000;Abbott et al., 2002). Using primary cultured bovine brain microvessel endothelial cells and the capillary-enriched fraction from bovine brain homogenates, reverse transcription-polymerase chain reaction analysis demonstrated the presence of MRP1, MRP4, MRP5, and MRP6 as well as low levels of MRP3, whereas MRP2 was absent (Zhang et al., 2000). However, using immunostaining of PGP and MRP2 in isolated capillaries from rat and pig brain, both multidrug transporters were localized to the luminal surface of the capillary endothelium (Miller et al., 2000). In rats, MRP1 is present in higher levels in astrocytes than in brain capillary endothelial cells (Decleves et al., 2000). Furthermore, high expression of MRP-1 is found in CP epithelial cells that form the BCB (Rao et al., 1999). The recent generation ofmrp gene knockout mice is providing information on the physiological functions of MRPs in these different localizations. Mice lacking an intact mrp1 gene have an altered response to inflammatory stimuli and show an increased toxic response to the anticancer drug etoposide, but are otherwise healthy (Borst et al., 2000). In mdr1a/mdr1b/mrp1 triple knockout mice, but not inmdr1a/mdr1b double knockout mice, etoposide levels in the CSF are markedly increased, indicating that MRP1 critically contributes to the permeability of the BCB (Wijnholds et al., 2000). In addition tomrp knockout mouse mutants, there is amrp2-deficient rat mutant that can be used to study physiological functions of MRP2 (Koopen et al., 1998). The role of MRPs in BBB permeability has been demonstrated by experiments in which inhibitors of MRPs, such as probenecid or MK-571, were shown to enhance drug penetration into the brain or to inhibit drug efflux from isolated brain endothelial cells (Gutmann et al., 1999; Potschka and Löscher, 2001a; Potschka et al., 2001; Sun et al., 2001).
The role of multidrug transporters such as PGP or MRPs in pharmacoresistance has been extensively studied in tumor cells that possess intrinsic or acquired cross-resistance to diverse chemotherapeutic agents (Tan et al., 2000; Litman et al., 2001). Drawing on parallels with resistance to cancer, some groups have begun to study the possibility that over-expression of multidrug transporters in normal tissues such as the BBB and BCB contributes to pharmacoresistance in other diseases, resulting in accumulating evidence that several multidrug transporters are over-expressed in the brain of patients with medically intractable epilepsy.
Over-Expression of Multidrug Transporters in Brain Tissue of Pharmacoresistant Patients with Epilepsy
Tishler et al. (1995) were the first to report that brain expression of MDR1, which encodes the multidrug transporter PGP in humans, is markedly increased in the majority of patients with medically intractable partial (mostly temporal lobe) epilepsy.MDR1 mRNA levels were determined by reverse transcription-polymerase chain reaction in brain specimens removed from patients during resective surgery for intractable epilepsy and compared with normal brain control specimens obtained from patients undergoing removal of arteriovenous malformations. In line with enhancedMDR1 expression in epileptogenic brain tissue, immunohistochemistry for PGP showed increased staining in capillary endothelium and astrocytes. Tishler et al. (1995) proposed that PGP may play a clinically significant role by limiting access of AEDs to the brain parenchyma, so that increased MDR1 expression may contribute to the refractoriness of seizures in patients with pharmacoresistant epilepsy. Subsequently, it was shown by other groups that, in addition to PGP, MRP1 and MRP2 are over-expressed in the brain tissue of pharmacoresistant patients (Table1A). Sisodiya et al. (1999) reported over-expression of PGP in glial cells of brain samples from patients with malformations of cortical development, which are often associated with medically intractable epilepsy. In a subsequent study, Sisodiya et al. (2001) found over-expression of MRP1 in dysplastic neurons, glia, and around vessels in surgically resected epileptogenic human brain tissue of patients with focal cortical dysplasia (FCD), an important malformation of cortical development causing refractory epilepsy. Furthermore, when determining PGP and MRP1 expression in three common causes of refractory epilepsy, namely dysembryoplastic neuroepithelial tumors, FCD, and hippocampal sclerosis, and comparing the expression in the abnormal, epileptogenic tissue with PGP and MRP1 expression in histologically normal adjacent tissue, Sisodiya et al. (2002) found over-expression of both PGP and MRP1 in reactive astrocytes in the epileptogenic tissue in all three conditions, and MRP1 over-expression in dysplastic neurons in FCD. The over-expression in astrocytes appeared most marked around blood vessels. In view of data indicating that the endothelial barrier function of the BBB is transiently disrupted during seizures (cf., Duncan and Todd, 1991), over-expression of multidrug transporters in glial end-feet covering the blood vessels may represent a “second barrier” under these conditions (Sisodiya et al., 2002). Sisodiya et al. (2002) proposed that over-expressed multidrug transporters lower the extracellular concentration of AEDs in the vicinity of the epileptogenic pathology and thereby render the epilepsy caused by these pathologies resistant to AED treatment. By using gene arrays to study mRNAs of multidrug transporters in endothelial cells isolated from surgically resected epileptic foci of patients with pharmacoresistant partial epilepsy, Janigro and colleagues determined increased expression of MDR1 and the gene encoding MRP2 (Dombrowski et al., 2001), indicating that over-expression of both PGP and MRP2 in the BBB may be involved in resistance to AEDs.
Over-expression of multidrug transporters in epilepsy and transport of antiepileptic drugs by multidrug transporters
An open question is whether the over-expression of PGP and MRPs in epileptogenic brain tissue of patients with pharmacoresistant epilepsy is a consequence of epilepsy, uncontrolled seizures, chronic treatment with AEDs, or combinations of these factors. Because pharmacoresistant patients have the same extent of neurotoxic side effects under AED treatment as patients who are controlled by AEDs, the over-expression of drug transporters in pharmacoresistant patients is most likely restricted to the epileptic focus or circuit. This is substantiated by the recent report of Sisodiya et al. (2002) in which over-expression of PGP and MRP1 was found in epileptogenic tissue but not adjacent normal tissue. In this respect, it is also interesting to note that in patients in whom the epileptic focus has been resected during epilepsy surgery—resulting in seizure control under treatment with AEDs—seizures may recur after cessation of AED treatment and become pharmacoresistant again, suggesting that a “secondary focus” has become activated and drug-resistant (Löscher, 2002). In rats, kainate-induced seizures have been found to transiently over-express PGP in astroglia and, less marked, capillary endothelial cells in the hippocampus (Zhang et al., 1999), indicating that seizures rather than epilepsy are responsible for over-expression of drug transporters. This could explain that one of the major predictors of pharmacoresistance is high seizure frequency prior to initiation of treatment (Regesta and Tanganelli, 1999). However, constitutive rather than induced or acquired over-expression of multidrug transporters has been reported in patients with malformations of cortical development (Sisodiya et al., 1999). In addition to intrinsic or acquired over-expression of multidrug transporters in the BBB or BCB of patients with epilepsy, functional polymorphisms of these transporters may play a role in pharmacoresistance (Kerb et al., 2001). Furthermore, over-expression and functional polymorphisms of multidrug transporters in patients with pharmacoresistant epilepsy need not necessarily be restricted to the brain, but could also occur in other tissues, such as the small intestine, where PGP is thought to form a barrier against entrance of drugs from the intestinal lumen into the bloodstream, thereby limiting their oral bioavailability (Fromm, 2000). In this respect, it is interesting to note that Lazarowski et al. (1999) have reported persistent subtherapeutic levels of AEDs (including phenytoin and phenobarbital) despite aggressive and continuous AED administration in a patient with refractory epilepsy associated with over-expression ofMDR1.
In view of the emerging evidence that multidrug transporters are over-expressed in epileptogenic brain tissue, particularly in capillary endothelial cells and astrocytes contributing to BBB permeability, it is of major clinical interest to evaluate whether AEDs are substrates for these transporters. Only then, over-expression of PGP or MRPs could critically contribute to pharmacoresistance in epilepsy.
Active Transport of Antiepileptic Drugs in the Blood-Brain and Blood-CSF Barriers
Based on the assumption that penetration of drugs from blood into brain and CSF depends mainly on the drugs' lipid solubility, drugs required to act within the brain, such as AEDs, have generally been made lipophilic. By studying the rate of entry of various AEDs from blood into the CSF of anesthetized dogs, Löscher and Frey (1984)found a significant correlation between penetration rate and lipid solubility, measured by organic solvent/buffer distribution ratios, whereas the extent of plasma protein binding and degree of ionization of AEDs were of minor importance for penetration rates. These data thus substantiated that lipid solubility plays the major role in determining the difference in rate of entry of AEDs, and that AEDs, as do most centrally active drugs, penetrate into the CSF by simple diffusion. However, one AED, valproate, did not fit into this scheme. Valproate, a branched medium-chain fatty acid, is almost completely ionized at physiologic pH, and its lipid solubility at this pH is therefore very low (Löscher and Frey, 1984). However, although it has been shown that drugs with such properties enter the CSF or brain very slowly if at all (Goldstein et al., 1974), valproate penetrated into the CSF and brain very rapidly (Löscher, 1982; Löscher and Frey, 1984). Indeed, valproate was the first AED for which an active transport in the BCB and BBB has been proposed (Frey and Löscher, 1978). In dogs, the rate of entry into CSF was markedly reduced at high drug concentrations, indicating saturation of transport (Frey and Löscher, 1978). The rate of entry of valproate into CSF as well as the CSF/plasma concentration ratio could be strikingly increased by probenecid, an inhibitor of organic acid transport carriers (Frey and Löscher, 1978). Subsequently, it was shown that probenecid also increased the concentration of valproate in the brain (Adkison et al., 1994). More recent animal studies revealed that the bidirectional movement of valproate across the BBB (and possibly also BCB) is mediated jointly by passive diffusion and carrier-mediated transport (Shen, 1999). The uptake of valproate from blood to brain is facilitated by a medium-chain fatty acid transporter, which accounts for two-thirds of the barrier permeability, whereas the mechanisms governing the efflux of valproate from the brain involve a probenecid-sensitive, active transport system at the brain capillary endothelium (Shen, 1999). Recent data from Huai-Yun et al. (1998) show that valproate is a substrate for MRPs in brain capillary endothelial cells, which raises the possibility that MRPs may serve as the efflux transporters of valproate and explains the previously described effects of probenecid on brain and CSF levels of valproate, because probenecid is an inhibitor of MRP1 and MRP2 (Hooijberg et al., 1999; Borst et al., 2000).
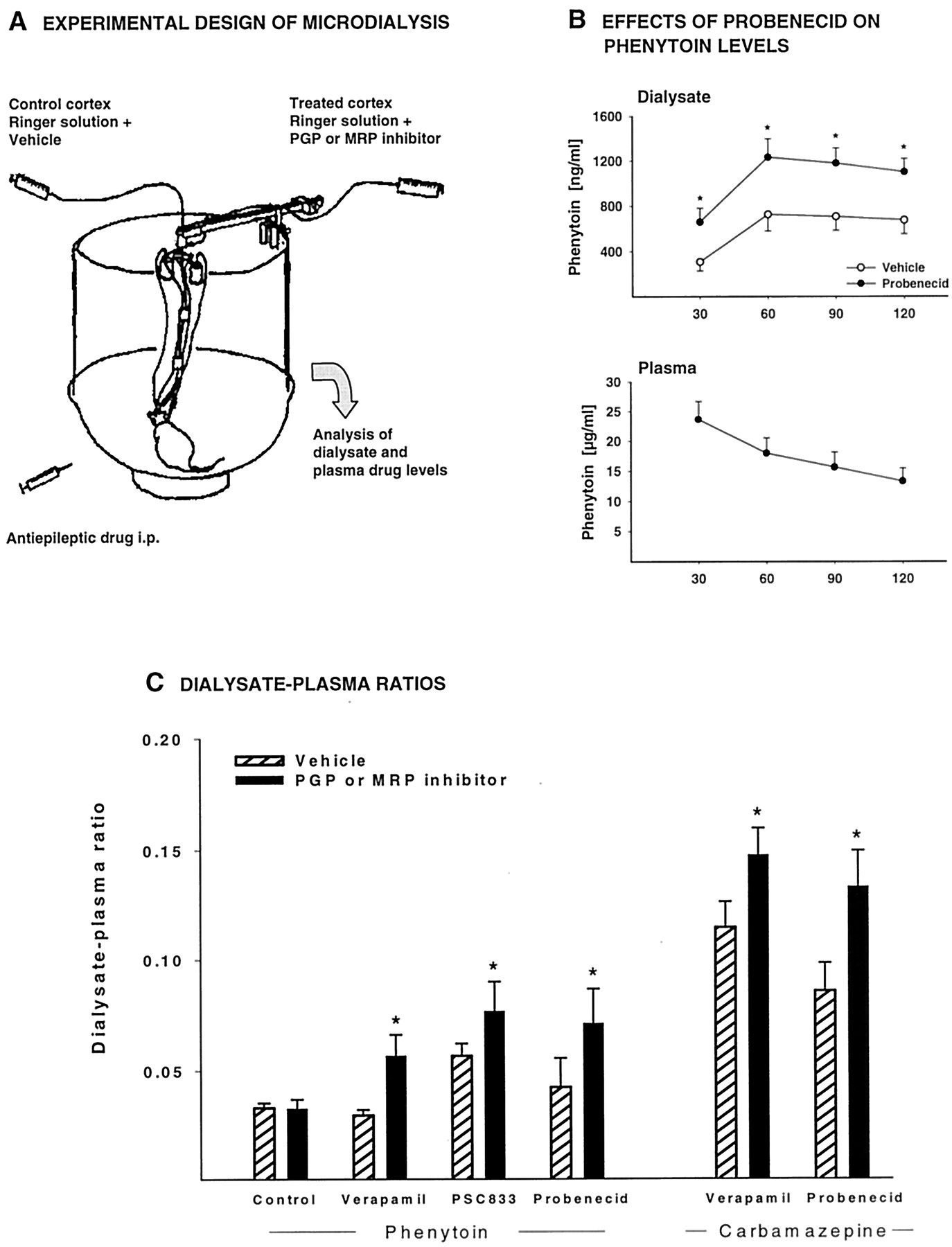
Except valproate, all other AEDs are highly lipid-soluble at physiologic pH, which makes them potential substrates for efflux carriers of the BBB, such as PGP and MRPs (Abbott et al., 2002). Indeed, there is increasing evidence that various major AEDs are substrates for one or more of these efflux carriers (Table 1B). At least three strategies are used in this respect. One is to evaluate whether the brain penetration of AEDs can be affected by PGP or MRP inhibitors; a second is to use cell lines that over-express PGP or MRPs; and a third is to study drug penetration into the brain ofmdr or mrp knockout mice. As described above, valproate was the first AED for which BCB and BBB transport by a probenecid-sensitive carrier, most likely MRP, has been reported. Using a brain microdialysis model in rats to study drug transport across the BBB (Fig. 2), we found that brain extracellular levels of phenytoin and carbamazepine can be significantly increased by PGP and MRP inhibitors, indicating that PGP and MRPs physiologically limit brain penetration of these major AEDs (Potschka and Löscher, 2001a,b; Potschka et al., 2001). Furthermore, by using the same model, we have preliminary evidence that phenobarbital, felbamate, and lamotrigine are substrates for PGP (H. Potschka, M. Fedrowitz, and W. Löscher, unpublished experiments). For the AED gabapentin, there is evidence for a saturable transport at the BBB, and BBB amino acid transport system-L has been suggested to be responsible for this transport (Luer et al., 1999). Furthermore, recent data from mdr1 knockout mice indicate that gabapentin is also a substrate for multidrug transporters (see below).

Intracerebral microdialysis in pharmacokinetic studies on drug transport across the BBB. A, the model used in our experiments on BBB transport of AEDs is presented. The experimental protocol used for this purpose is similar to the protocol described byBurgio et al. (1998) for studying the involvement of PGP in the control of brain distribution of the chemotherapeutic agent etoposide. In short, rats are implanted with two microdialysis probes in the left and right motor cortices. One probe is infused with an aqueous Ringer's solution containing an inhibitor of either PGP or MRP and the other probe with Ringer's solution and the vehicle used for dissolving the inhibitor. The AED is then injected i.p., and plasma and dialysate concentrations are repeatedly determined in conscious, freely moving rats. Because only one cortex is treated with a PGP or MRP inhibitor, the vehicle-treated cortex serves as control site in each individual rat. B, data of a typical experiment in a group of five rats are shown, using the major AED phenytoin (50 mg/kg i.p.) and the MRP1/2 inhibitor probenecid. Probenecid significantly increased the extracellular (dialysate) level of phenytoin in the treated cortex (★,P < 0.05 versus untreated cortex), without affecting the plasma kinetics of phenytoin. C, the effects of PGP inhibitors (verapamil, PSC 833) and the MRP1/2 inhibitor probenecid on the ratio between drug levels in dialysate and plasma are compared for two AEDs, phenytoin and carbamazepine, in groups of 5 to 10 rats. Ratios were calculated for 0 to 120 min after AED administration. An additional group (“control”) received phenytoin but no intracerebral administration of a PGP or MRP inhibitor. For each group, significant differences between vehicle- and inhibitor-treated cortex are indicated by an asterisk (P < 0.05). The data demonstrate that intracerebral administration of PGP and MRP inhibitors significantly increases dialysate levels of phenytoin and carbamazepine, indicating that PGP and MRPs are involved in the regulation of extracellular levels of these AEDs in the brain. For further details see Potschka and Löscher (2001a,b) and Potschka et al. (2001).
With respect to the use of cell lines to study AED transport, Tishler et al. (1995) found that intracellular phenytoin levels in aMDR1-expressing neuroectodermal cell line were only one-fourth that in MDR1-negative cells, suggesting that PGP significantly contributes to cell export of phenytoin. In a kidney epithelial cell line transfected with mdr1a cDNA, phenytoin was transported, which could be blocked by the PGP inhibitor PSC 833 (valspodar; Schinkel et al., 1996). The transport of carbamazepine was studied in Caco-2 cells, an in vitro model of the intestinal epithelium known to express high PGP levels (Owen et al., 2001). In these cells, the transport of carbamazepine was PGP-independent, and was not affected by PSC 833 (Owen et al., 2001). In human colon carcinoma cells, phenobarbital and, to a much lesser extent, phenytoin were found to up-regulate PGP, a phenomenon described for several substrates of PGP (Schuetz et al., 1996).
In mdr1 knockout mice, in which PGP is absent in the BBB, brain levels of phenytoin and carbamazepine were reported to be not different from wild-type mice (Schinkel et al., 1996; Owen et al., 2001). However, in another study in mdr1 knockout mice, the brain plasma/concentration ratio for carbamazepine was found to be significantly higher in knockout mice than in wild-type controls (Sills and Kwan, 2001). Furthermore, significant increases in brain levels were found for topiramate, lamotrigine, and gabapentin inmdr1 knockout mice, although no significant differences to controls were seen for phenobarbital, phenytoin, valproate, and vigabatrin (Sills and Kwan, 2001). However, use of knockout mice is limited in the study of drug resistance because of the redundancy of the transporters: another transport protein may take over the function of one that has been knocked out (Schinkel, 1999). Thus, failure of knockout to affect AED kinetics cannot be taken to prove that the protein knocked out does not transport AEDs. Furthermore, considering the relatively small increases in extracellular brain levels of carbamazepine and phenytoin by PGP or MRP inhibition in rats (see Fig.2), such increases may be missed when these AEDs are determined in whole brain tissue, as was done in the studies using knockout mice. A further point when considering different results on AED transport from different model systems are polymorphisms in the genes encoding PGP and MRPs, resulting in functional alterations in these drug transporters (Kerb et al., 2001).
Although there are some inconsistencies when studying transport of AEDs by PGP or MRPs in different model systems, the emerging impression is that a number of major AEDs are subject to active transport by PGP or MRPs in the BBB or BCB. Although the available data indicate that AEDs are only relatively weak substrates for multidrug transporters under normal conditions, over-expression of such transporters could significantly reduce brain levels of these drugs and thereby critically contribute to pharmacoresistance in epilepsy.
However, although over-expression or up-regulation of efflux transporters at the BBB would be a plausible mechanism for drug-resistant epilepsy, this hypothesis has not yet been tested directly to any significant extent. It is difficult to know whether the over-expression of multidrug transporters in epileptogenic brain tissue is a major cause of resistance, or merely a secondary effect of disease, i.e., an epiphenomenon. There are at least two direct approaches to test the hypothesis. One is to determine whether brain levels of AEDs are decreased in epileptogenic brain tissue with over-expressed transporters; another is to evaluate whether coadministration of PGP or MRP inhibitors can reverse pharmacoresistance in epilepsy. In kindled rats, which are one of the few chronic animal models of drug-resistant partial epilepsy (Löscher, 1997), we recently found lower extracellular levels of phenytoin in brain regions involved in seizure generation compared with nonkindled controls (Potschka and Löscher, 2002), which would be in line with the concept that seizure-induced over-expression of drug transporters limits access of AEDs to the brain parenchyma, thereby contributing to drug resistance in epilepsy.
Consequences for Pharmacotherapy of Epilepsy and Drug Development
Provided that over-expression of multidrug transporters is involved in pharmacoresistance in epilepsy, this would allow novel options for treatment of refractory epilepsy, such as addition of an inhibitor of multidrug transporters to current treatment with AEDs. We currently test whether coadministration of AEDs with PGP or MRP inhibitors in animal models of temporal lobe epilepsy such as kindling results in enhanced anticonvulsant activity. Furthermore, as a “proof of principle”, we plan to evaluate whether pharmacoresistance to AEDs can be overcome by adding a PGP or MRP inhibitor, using AED-resistant subgroups of kindled rats (cf., Löscher, 1997). With respect to PGP inhibitors that can be used for such experiments, there are currently three generations of inhibitors (Tan et al., 2000). The first generation includes drugs such as cyclosporin A or verapamil, which are not selective but exert several other effects apart from PGP inhibition. The second generation, e.g., the cyclosporin A analog PSC 833 (valspodar), is much more selective, but exerts inhibitory effects on drug metabolism by blocking cytochrome P450 3A4, which is also the case with several of the first generation PGP inhibitors, e.g., verapamil. The third generation, e.g., the cyclopropyldibenzosuberane derivative LY 335979, VX 710 (biricodar), or the diarylimidazole derivative OC 144-093, selectively inhibits PGP without interfering with drug metabolism. The second and third generation drugs, which have been developed for treatment of multidrug resistant cancer, are well tolerated in humans, ataxia being the main adverse effect. Inhibitors of MRP include probenecid and the more selective MK-571 and LY402913 (Sun et al., 2001). There are also unspecific inhibitors, such as sodium cyanide, which block all types of ATP-dependent multidrug transporters, and markedly increase dialysate levels of phenytoin in the rat model shown in Fig. 1 (Potschka and Löscher, 2001b), but their use is limited by toxicity.
The only PGP inhibitors that have been clinically evaluated in combination with AEDs in patients with epilepsy are calcium channel blockers such as verapamil, nifedipine, or diltiazem (Löscher and Schmidt, 1994). Verapamil and diltiazem increased the plasma concentrations of carbamazepine (probably by inhibiting its CYP3A4-mediated metabolism) and caused unacceptable neurotoxicity, but encouraging clinical observations were reported for add-on treatment with nifedipine in patients with refractory partial seizures (Löscher and Schmidt, 1994). However, because calcium channel antagonists exert anticonvulsant activity of their own and inhibit CYP3A4, it is not possible to judge whether the favorable effect of combinations of nifedipine and AEDs was due to inhibition of PGP, inhibition of CYP3A4, or blockade of calcium channels. Definite conclusions about the role of PGP and MRPs in pharmacoresistant epilepsy have to await animal experiments and clinical trials with more selective inhibitors.
With respect to development of new AEDs, drugs not transported by multidrug transporters expressed in the BBB could have advantages toward available AEDs in patients with pharmacoresistant epilepsy and focal over-expression of such transporters. PGP assays to identify drugs that are not PGP substrates are already routinely used in drug development in the pharmaceutical industry, but this does not exclude that other transporters such as MRPs accept such drugs as substrates. However, when searching lipid-soluble drugs that are poor or no substrates for PGP or MRP, it should be noted that because multidrug transporters are thought to protect a number of organs from intoxication by xenobiotics (Leveille-Webster and Arias, 1995), lipophilic drugs that are not restricted in tissue distribution by such transporters may have a low therapeutic margin.
Conclusions
Multidrug transporters such as PGP and MRPs are important gatekeepers in the BBB and BCB, and there is increasing evidence that over-expression of such multidrug transporters may be involved in the generation of pharmacoresistance in epileptic patients. If so, inhibitors of these drug transporters may prove useful in pharmacoresistant epilepsy. Inhibitors of PGP and, more recently, MRPs are currently clinically evaluated for reversal or prevention of intrinsic and acquired multidrug resistance in human cancer (Litman et al., 2001) and may soon become available for clinical trials on adjunctive treatment in refractory epilepsy, although the adverse effects of PGP or MRP inhibition need careful consideration. As with every new therapeutic option, overly high expectations should be avoided, particularly because there is as yet no direct proof that over-expression of multidrug transporters is a possible cause of drug resistance in the treatment of epilepsy. In addition, other mechanisms of pharmacoresistance should be identified, because it is likely that different factors underlie multidrug resistance in epilepsy. Further research of the basic mechanisms of drug resistance in epilepsy should help to identify new approaches to the rational treatment of epilepsy, for example by design of AEDs that are no targets for brain-expressed resistance mechanisms.
Acknowledgments
We thank Dr. Manuela Gernert for help with the illustrations.
Footnotes
-
The epilepsy research program of the authors and this study are supported by grants from the Deutsche Forschungsgemeinschaft (Bonn, Germany).
- Abbreviations:
- AED
- antiepileptic drug
- BBB
- blood-brain barrier
- CP
- choroid plexus
- CSF
- cerebrospinal fluid
- BCB
- blood-CSF barrier
- FCD
- focal cortical dysplasia
- MRP
- multidrug resistance-associated protein
- PGP
- P-glycoprotein
- Received October 30, 2001.
- Accepted December 20, 2001.
- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Pharmacoresistance to Antiepileptic Drugs in Patients with Epilepsy
- Multidrug Transporters in the Blood-Brain and Blood-Cerebrospinal Fluid (CSF) Barriers
- Over-Expression of Multidrug Transporters in Brain Tissue of Pharmacoresistant Patients with Epilepsy
- Active Transport of Antiepileptic Drugs in the Blood-Brain and Blood-CSF Barriers
- Consequences for Pharmacotherapy of Epilepsy and Drug Development
- Conclusions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters