


Abstract
Evidence from postmortem analysis implicates the involvement of microglia in the neurodegenerative process of several degenerative neurological diseases, including Alzheimer's disease and Parkinson's disease. It remains to be determined, however, whether microglial activation plays a role in the initiation stage of disease progression or occurs merely as a response to neuronal death. Activated microglia secrete a variety of proinflammatory and neurotoxic factors that are believed to induce and/or exacerbate neurodegeneration. In this article, we summarize recent advances on the study of the role of microglia based on findings from animal and cell culture models in the pathogenesis of neurodegenerative diseases, with particular emphasis on Parkinson's disease. In addition, we also discuss novel approaches to potential therapeutic strategies.
Microglia
Microglia are considered the resident immune cells of the central nervous system (CNS). Since the initial comprehensive description of microglia by del Rio-Hortega in 1932, the exact origin of microglia remains the subject of debate. Numerous studies in the last three decades, however, have generally supported the view that microglia derive from mesodermal precursor cells of possibly hematopoietic lineage that enter the brain during the embryonic and early postnatal phases of development (for reviews, see Barron, 1995; Cuadros and Navascues, 1998). During brain remodeling and maturation, microglia are believed to assist in the clearance of cells deemed for elimination through programmed cell death. In the mature brain and under physiological conditions, resting microglia adopt the characteristic ramified morphological appearance and serve the role of immune surveillance and host defense. Microglia, however, are particularly sensitive to changes in their microenvironment and readily become activated in response to infection or injury. Activated microglia up-regulate a variety of surface receptors, including the major histocompatibility complex and complement receptors. They also undergo dramatic morphological changes from resting ramified cells to activated amoeboid microglia (Kreutzberg, 1996).
Besides morphological changes and surface molecule up-regulation, activated microglia secrete a host of soluble factors. A number of these factors, such as the glia-derived neurotrophic factor, are potentially beneficial to the survival of neurons, reminiscence of the neuroprotective role played by activated astrocytes, another major type of glial cells in the brain (Aloisi, 1999). The majority of factors produced by activated microglia, however, are proinflammatory and neurotoxic. These include the cytokines tumor necrosis factor-α (TNFα) and interleukin-1β (IL-1β), free radicals such as nitric oxide (NO) and superoxide, fatty acid metabolites such as eicosanoids, and quinolinic acid. Studies using cell culture and animal models have demonstrated that excessive quantities of individual factors produced by activated microglia can be deleterious to neurons (Boje and Arora, 1992; Chao et al., 1992; McGuire et al., 2001). Furthermore, individual factors often work in concert to induce neurodegeneration. For example,Chao et al. (1995) reported that the combination of IL-1β and TNFα, but not either cytokine alone, induced the degeneration of cortical neurons. Jeohn and colleagues (1998) have shown that the combination of IL-1β, TNFα, and interferon-γ work in synergy to induce degeneration of cortical neurons. Recently, Xie et al. (2002)showed that peroxynitrite, possibly a product of superoxide and NO, is a major mediator of neurotoxicity induced by lipopolysaccharide (LPS) or β-amyloid peptide (1-42).
Evidence for the Involvement of Microglia in Neurodegenerative Diseases
The involvement of microglial activation in the pathogenesis of several neurodegenerative diseases was initially postulated based on the postmortem analysis of the brains of patients with Alzheimer's disease (AD) and Parkinson's disease (PD). For instance, reactive microglia were found to colocalize with neuritic plaques in the cortical region of AD brains (Rogers et al., 1988). In PD brains, large numbers of human leukocyte antigen (HLA-DR)-positive reactive microglia were found in the substantia nigra (SN), a region in which the degeneration of dopaminergic neurons was most prominent (McGeer et al., 1988). In addition to AD and PD, results from both in vivo and in vitro studies have since established an association of microglial activation with the pathogenesis of human immunodeficiency virus (HIV) acquired immunodeficiency syndrome dementia complex, amyotrophic lateral sclerosis, multiple sclerosis, and prion-related diseases (Dickson et al., 1993; Raine, 1994; Brown, 2001).
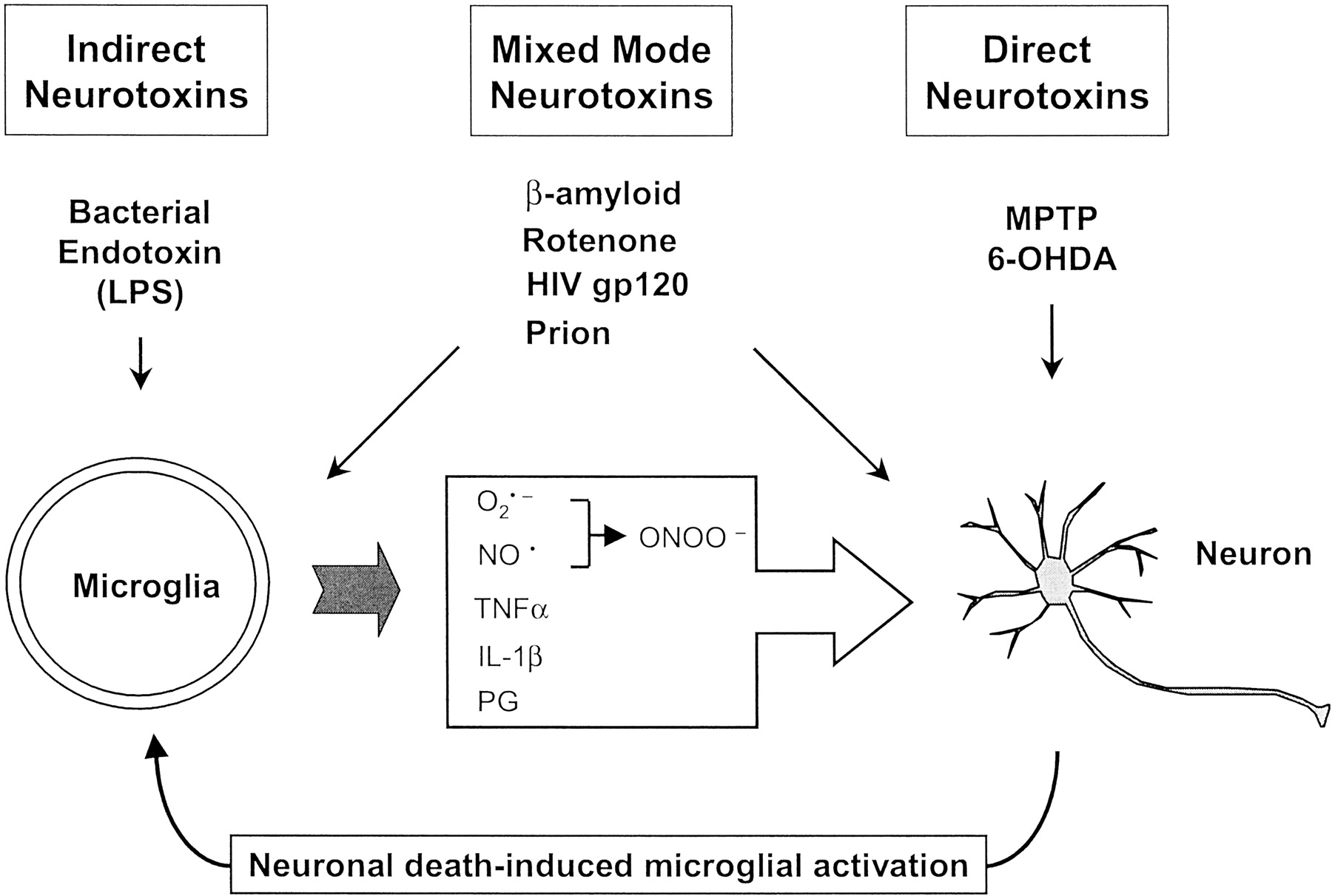
Multiple Pathways Leading to Microglial Activation
It is now generally accepted that microglia contribute to the neurodegenerative process through the release of a variety of neurotoxic factors that exacerbate the degeneration of neurons. It remains to be determined, however, what triggers microglial activation in these various disorders. Important clues relevant to understanding the pathogenesis of degenerative neurological disorders can be obtained by comparing the mode of action of a wide spectrum of potentially neurotoxic agents (Fig. 1). At one end of the spectrum are those agents that appear to be totally incapable of directly killing neurons. One of the best-studied agents in this group may be the bacterial cell wall endotoxin LPS. LPS is a widely used and powerful tool for the activation of microglia and of peripheral immune cells. Although LPS has no known direct toxic effect on neurons, it activates microglia to release a host of neurotoxic factors to induce neuronal death (Bronstein et al., 1995; Araki et al., 2001; Liu et al., 2002c). In contrast to the action of LPS, certain agents are known to have a direct neurotoxic effect. Two experimental neurotoxins for dopaminergic neurons, namely 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), are good examples. Direct damage to neurons by these agents causes reactive gliosis. Activation of microglia in turn exacerbates the neurodegenerative process. For example, mice lacking inducible nitric oxide synthase activity are resistant to MPTP-induced lesions and inhibition of microglial activation reduces MPTP neurotoxicity (Itzhak et al., 1999; Liberatore et al., 1999;Dehmer et al., 2000; Du et al., 2001; Wu et al., 2002). In the striatum and SN of 6-OHDA lesioned rat brains, prominent microglial activation was detectable weeks after the lesion (Cicchetti et al., 2002).

A simplified classification of three types of potentially neurotoxic agents.
In addition to the direct and indirect toxins mentioned above, it is interesting to note that another group of agents that are known to be associated with various neurodegenerative diseases exhibit a “mixed mode” mechanism of neurotoxicity. These include β-amyloid peptides (Aβ), HIV coat protein gp120, prion protein-derived peptides, and the pesticide rotenone (Fig. 1). Previous reports have generally attributed their neurotoxicity to a direct impact on neurons. Recent studies from several laboratories, however, have revealed that activation of microglia and the subsequent release of neurotoxic factors contribute to their neurotoxicity. For example, the potency of Aβ (1-42)-induced neurotoxicity on cultured cortical and mesencephalic neurons increased by severalfold in the presence of microglia (Qin et al., 2002). The enhanced neurotoxicity of Aβ (1-42) was attributed primarily to the activation of microglia and subsequent release of the superoxide free radical. In the case of rotenone, which was initially thought to damage dopaminergic neurons by inhibiting mitochondrial complex I activity, the presence of microglia significantly enhanced its neurotoxicity, and the generation of oxygen free radicals appeared to underlie this increased toxicity (Gao et al., 2002a). Similarly, the destruction of neurons by HIV gp120 and prion proteins also involves the participation of activated microglia (Brown, 2001). Therefore, it appears highly probable that the overall neurotoxic effect of these mixed mode toxins includes elements of both direct neurotoxicity and indirect toxicity through the activation of microglia. It now seems clear that microglial activation is involved in the pathogenic process of multiple forms of degenerative neurological diseases. It still remains to be determined whether microglial activation plays a role in the earliest stages of disease development, however.
Besides exposure to neurotoxins, neuronal damage as a consequence of ischemic and mechanical injuries elicits microglial activation and constitutes secondary neuronal loss (McMillian et al., 1994). Neurons, through mechanisms that include cell-cell contact via cell adhesion molecules, appear to suppress the reactivity of microglia (Chang et al., 2000a). A reduction in the ratio of neurons to microglia in culture was shown to reduce the modulatory effect of neurons on glia, resulting in increased glial reactivity to LPS (Chang et al., 2001).
Microglial Activation in PD: Relevance to Etiology
PD is characterized by a progressive and selective destruction of the nigrostriatal dopaminergic system that is important in the regulation of body movements. Except for a small faction (<5%) of mostly early onset and familial PD, the clinical symptoms of the majority as well as of sporadic cases of PD occur late in life and progress over decades (Olanow and Tatton, 1999).
The detection of elevated levels of proinflammatory cytokines and evidence of oxidative stress-mediated damage in postmortem PD brains has lent strong support to the notion of microglial involvement in the degenerative process. More compelling, epidemiological studies and case reports seem to indicate a positive correlation between early-life brain injuries and late development of PD, implying that inflammation in the brain, and specifically microglial activation, plays a critical role in the early stage(s) of the pathogenesis of this disorder (McGeer et al., 1988). First, occurrence of antecedent traumatic brain injury appears to increase the incidence of late-life development of PD, AD, or dementia in general (Factor et al., 1988). Second, it has long been speculated that populations of people exposed to certain viruses or other infectious agents have an increased probability of developing postencephalitic Parkinsonism, sometimes several decades later (Casals et al., 1998). Third, intrauterine fetal brain inflammation following exposure to viruses or endotoxins may play a role in the late development of PD (Ling et al., 2002). The SN region of the brain is particularly rich in microglia (Lawson et al., 1990; Kim et al., 2000). In addition, dopaminergic neurons in the SN are known to have a reduced antioxidant capacity, evidenced by a reduced level of the intracellular glutathione, rendering them uniquely vulnerable to a variety of insults, including oxidative stress (Greenamyre et al., 1999). Therefore, it is logical to infer that activation of microglia, as a consequence of neuronal injury or infection, represents a risk factor that may trigger the onset of a cascade of events leading to a progressive degeneration of dopaminergic neurons.
Inflammogen-Induced Microglial Activation and Dopaminergic Neurodegeneration
To test this hypothesis, an inflammation-mediated rat model of PD was established in our laboratory by supranigral and continuous infusion of nanogram quantities of an inflammogen (LPS) for 2 weeks (Gao et al., 2002b). Maximal activation of microglia in the SN occurred between 1 to 2 weeks after the start of LPS infusion. Degeneration of nigral dopaminergic neurons, however, did not begin until 3 to 4 weeks after the occurrence of peak microglial activation. Furthermore, the delayed degeneration of nigral neurons was both selective to dopaminergic neurons and progressive. Ten weeks after the initiation of LPS infusion, a 70% loss of nigral dopaminergic neurons was observed. In a parallel in vitro cell culture model of PD, the factors involved in LPS-induced neurodegeneration were identified. Depending on the concentrations of LPS used to stimulate the mesencephalic neuron-glia culture, the profiles of microglia-produced neurotoxic factors were different. At 3 to 10 ng/ml LPS, significant quantities of NO, TNFα, and superoxide were produced. In contrast, at 0.1 to 1 ng/ml LPS, only significant quantities of superoxide, but not NO and TNFα, were detected. Furthermore, neutralization of the reactivity of superoxide or inhibition of superoxide production with inhibitors of microglial NADPH oxidase afforded significant neuroprotection. These results demonstrate that free radicals generated of by LPS-activated microglia are a primary contributor to the degeneration of dopaminergic neurons. This observation is consistent with the assumption that dopaminergic neurons are particularly susceptible to oxidative damage (Greenamyre et al., 1999). A single injection of a combination of TNFα, IL-1β, and interferon-γ to the SN was found to be insufficient to induce significant neurodegeneration (Castano et al., 2002). Results obtained from these models of inflammation-mediated dopaminergic neurodegeneration demonstrate that microglial activation induced by chronic exposure to low levels of bacterial endotoxin is capable of inducing a delayed and selective degeneration of nigral dopaminergic neurons.
The chronic LPS infusion-induced PD rodent model differs from the single intranigral injection-induced acute lesion model in several important aspects (Fig. 2). First, neurodegeneration induced by a single bolus application of microgram levels of LPS occurred within a few days (Castano et al., 1998; Liu et al., 2000d; Lu et al., 2000; Iravani et al., 2002). In contrast, neurodegeneration induced by chronic infusion of nanogram levels of LPS progressed over weeks (Gao et al., 2002b). Second, in the acute model, neurodegeneration was not limited to nigral dopaminergic neurons because other neurons such as γ-aminobutyric acid-containing neurons were also damaged. In contrast, in the chronic model, neurodegeneration was selective for nigral dopaminergic neurons. Third and perhaps most important, in the acute model, LPS-induced microglial activation occurred either at the same time or immediately preceding apparent neurodegeneration. By contrast, LPS-induced neurodegeneration in the infusion model occurred in a delayed fashion by starting weeks after the apex of microglial activation. Therefore, the chronic infusion model of LPS-induced dopaminergic lesions may be a more suitable tool than the acute model to further analyze the relationship between microglial activation and dopaminergic neurodegeneration.

Comparison of the single injection (acute) and infusion (chronic) rodent PD models of LPS-induced dopaminergic neurodegeneration. Microglial activation indicates the activation status of nigral microglia immunostained with the antibody against the complement CR3 receptor (OX-42). Neuronal loss refers to the loss of nigral tyrosine hydroxylase-immunoreactive neurons (i.e., dopaminergic neurons).
Besides supranigral infusion of LPS into adult rodent brains, the impact of in utero exposure to LPS on the induction of dopaminergic lesions, under the clinical context of bacterial vaginosis, has also been examined (Ling et al., 2002). Administration of LPS during pregnancy leads to a reduced striatal dopamine content and nigral dopaminergic neurons in offspring 21 days after birth. Finally, it will be informative to examine the influence of systemic inflammation (e.g., septic shock), as opposed to locally invoked inflammation, on the nigrostriatal dopaminergic system.
It should be emphasized that PD and AD are age-related degenerative neurological disorders. Microglial activation certainly plays an important role in the pathogenic, and perhaps even the initiation, stage of PD. Inflammation in the brain as a consequence of either exposure to infectious agents or neuronal injuries may represent only one of many factors in the etiology of the disorder. PD may prove to be the consequence of a complex interplay among multiple factors that include genetic predisposition, exposure to environmental toxins and the unique vulnerability of SN dopaminergic neurons.
It should also be pointed that astrocytes also play an important role in the pathogenic process of neurodegeneration. Reactive astrogliosis is frequently observed in the lesioned regions in the brains of patients with AD and PD (Aloisi, 1999). Interestingly, reactive astrogliosis usually lags behind the occurrence of microgliosis, as demonstrated in the MPTP-induced degeneration of nigral dopaminergic neurons in mice (Liberatore et al., 1999). Besides producing proinflammatory and neurotoxic factors such as NO and IL-1β, astrocytes can secrete neurotrophic factors to support the survival of neurons (Aloisi, 1999).
Neuroprotective Properties of Naloxone
The important role that microglial activation plays in inflammation-mediated neurodegeneration, and potentially in the pathogenesis of PD as well as AD, prompted us to speculate that inhibition of microglial activation would be neuroprotective. In the course of studying the effect of the endogenous opioids on glial cell activity in the brain, we discovered that the opioid receptor antagonist naloxone was capable of reducing the LPS-stimulated production of cytokines and nitric oxide in glial cultures (Das et al., 1995; Kong et al., 1997).
Naloxone is a nonselective antagonist of the G-protein-linked classic opioid receptors that are widely expressed on cells of the central nervous system (CNS) as well as the peripheral systems. Interaction of endogenous opioid peptides (enkephalins, dynorphins, and β-endorphins) with their respective opioid receptors results in the modulation of a variety of cellular activities, including the nociceptive/analgesic effects, respiration, ion channel activity, and immune responses. Binding of naloxone to opioid receptors is stereospecific, where only the (−)-naloxone isomer is effective. The affinity of the enantiomer (+)-naloxone for classical opioid receptors, however, is 3 to 4 orders of magnitude lower than that of (−)-naloxone (Fig. 3).

Potential mechanism of action for the neuroprotective effect of naloxone stereoisomers against neurodegeneration induced by LPS or Aβ (1-42).
The discovery that (−)-naloxone was able to attenuate LPS-induced microglial activation raised the possibility that naloxone might be neuroprotective in experimental models of inflammation-mediated neurodegenerative diseases. In the rat mesencephalic neuron-glia culture system, (−)-naloxone (1 μM) significantly attenuated the degeneration of dopaminergic neurons (Liu et al., 2000a). More importantly, the ineffective opioid receptor antagonist (+)-naloxone was also effective. The neuroprotective effect of naloxone was confirmed in the inflammation-mediated rodent PD model (Liu et al., 2000d; Lu et al., 2000). Furthermore, both naloxone isomers were equally effective in the attenuation of the LPS-induced nigral dopaminergic neurodegeneration (Liu et al., 2000d).
The neuroprotective effect of naloxone appeared to be unrelated to the opioid system since both the opioid receptor antagonist (−)-naloxone and the receptor binding ineffective (+)-naloxone were equally effective in affording neuroprotection. Since microglial activation and the release of neurotoxic factors underlie the inflammation-mediated neurodegeneration, the effect of naloxone isomers on microglial activation was investigated. In the acute model for inflammation-mediated PD, infusion of LPS into the nigral area induced microglia to undergo rapid activation that is detectable within the first 24 h. In rats receiving systemic infusion (via an osmotic minipump) of either (−)-naloxone or (+)-naloxone, LPS-induced nigral microglial activation was significantly reduced. In mesencephalic neuron-glia cultures, the naloxone stereoisomers inhibited LPS-induced microglial activation and production of TNFα, IL-1β, NO, and superoxide free radicals (Liu et al., 2000a). Among the factors released by LPS-activated microglia, inhibition of superoxide generation by naloxone isomers was most pronounced (Chang et al., 2000b; Liu et al., 2000a). In addition to mesencephalic neurons, LPS-induced activation of cortical microglia and degeneration of cortical neurons in neuron-glia cocultures were also attenuated by naloxone isomers (Liu et al., 2000b).
Furthermore, the neuroprotective effect of naloxone isomers was not limited to LPS-induced neurodegeneration. Degeneration of cortical or mesencephalic neurons in neuron-glia cultures induced by treatment with Aβ (1-42) was also significantly reduced (Liu et al., 2002c). Neurodegeneration under those conditions was found to involve the activation of microglia and, specifically, the generation of superoxide free radical (Qin et al., 2002). The potential mechanism of action responsible for neuroprotective effect of naloxone was attributed to the inhibition of the production of superoxide in Aβ (1-42)-activated microglia. Furthermore, naloxone methiodide, a partial opioid antagonist that carries a charged group, was also capable of suppressing Aβ (1-42)-induced superoxide generation and affording neurodegeneration, suggesting that the site of action for naloxone was at the cell surface. Since LPS and β-amyloid peptides interact with distinct cell surface molecules for signal transduction (Hoffmann et al., 1999; Tan et al., 1999), naloxone most likely intercepts a convergent point downstream of the binding of LPS or Aβ (1-42) to the cell surface (Fig. 3).
The unique property of naloxone isomers to preferentially inhibit the production of superoxide free radicals in microglia and to afford neuroprotection supports the theory that reactive oxygen species (ROS) are a major contributor to neurodegeneration. Furthermore, studies on LPS-stimulated macrophages have demonstrated that generation of ROS is an upstream event serving to regulate the production of other proinflammatory factors such as TNFα and IL-1β (Sanlioglu et al., 2001; Hsu and Wen, 2002). In addition, it has been demonstrated the expression of IL-1β in an Aβ peptide-stimulated microglial cell line is modulated by ROS (Kang et al., 2001). Hence, it is possible that in immune cells of both the central and peripheral systems, generation of ROS is a very early response that facilitates the expression of genes for proinflammatory cytokines. Conversely, compared with conventional therapeutic strategies that inhibit the production of individual cytotoxic factors, inhibition of ROS generation by agents such as naloxone could be a highly effective approach. It is noteworthy that at the concentrations tested (0.1–10 μM), neither of the naloxone stereoisomer had any effect on the cytochromec-reducing potential of superoxide generated by the xanthine/xanthine oxidase system (B. Liu and J.-S. Hong, unpublished observations). Hence, it is highly unlikely that naloxone works as a scavenger for free radicals.
A review of the literature indicates (−)-naloxone has been tested in both animal models and clinical trials to treat a wide range of disease conditions, including drug abuse, alcohol addiction, eating disorders, spinal cord injury, shock, and cerebral and cardiac ischemia (for a review, see Liu et al., 2002c). Although the efficacy of naloxone in treating diseases such as drug abuse is clearly related to the opioid receptor system, the mechanisms of action responsible for the efficacy of naloxone in the majority of the aforementioned diseases are far from clear. The progression of some, if not all, of those diseases involves inflammation. For example, activation of immune cells in response to initial tissue injury and microbial infections has been associated with the pathogenesis of ischemic brain as well as myocardial tissue injuries (Vallance et al., 1997; Stoll et al., 1998). A key component of the inflammatory process is the generation of ROS such as superoxide by microglia in the CNS and by neutrophils and macrophages in the peripheral system. Therefore, inhibition of superoxide generation by naloxone in both the CNS and the peripheral system (Simpkins et al., 1985) may be part of the mechanism of action responsible for the observed protective effects. Since the (+)-naloxone isomer has little activity as an opioid receptor antagonist but is an effective inhibitor of immune cell activation, it may have a broadly applicable protective effect against oxidative stress- and inflammation-mediated damage to vulnerable cells of the CNS or peripheral system.
Neuroprotective Effect of Other Anti-Inflammatory Agents
Over the years, certain steroids have been tested for their neuroprotective effects. One of the most studied anti-inflammatory steroids is the glucocorticoid dexamethasone. In cell culture and animal models, dexamethasone has been found to decrease the production of proinflammatory cytokines and protect nigral dopaminergic neurons against LPS-induced degeneration (Hoozemans et al., 2001; Castano et al., 2002). The potential side effects of steroids limit its long-term clinical usage. The discovery of the inducible form of cyclooxygenase (COX-2) has further prompted the examination of nonsteroidal anti-inflammatory drugs as effective neuroprotective agents. Cyclooxygenase inhibitors in general have shown promise in the treatment o neurodegenerative diseases (Lipsky, 1999).
Conclusion
Microglial activation may play a pivotal role in the initiation and progression of several neurodegenerative diseases. Inhibition of microglial activation, therefore, would be an effective therapeutic approach to alleviating the progression of diseases such as AD and PD. Naloxone, especially (+)-naloxone, is a useful candidate for such as approach. Continued exploration of the mechanism(s) of action underlying the involvement of microglia in neurodegeneration and the inhibitory effect of naloxone on microglial activation is highly warranted.
Acknowledgments
We thank Drs. Cynthia Cooper and Meryl Patton for reading the manuscript. Due to space limitation, the authors express regret for not being able to cite all of the relevant publications.
Footnotes
-
DOI: 10.1124/jpet.102.035048
- Abbreviations:
- CNS
- central nervous system
- TNFα
- tumor necrosis factor-α
- IL-1β
- interleukin-1β
- NO
- nitric oxide
- LPS
- lipopolysaccharide
- AD
- Alzheimer's disease
- PD
- Parkinson's disease
- SN
- substantia nigra
- HIV
- human immunodeficiency virus
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA
- 6-hydroxydopamine
- Aβ
- beta amyloid peptide
- ROS
- reactive oxygen species
- Received June 6, 2002.
- Accepted August 13, 2002.
- U.S. Government
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Microglia
- Evidence for the Involvement of Microglia in Neurodegenerative Diseases
- Multiple Pathways Leading to Microglial Activation
- Microglial Activation in PD: Relevance to Etiology
- Inflammogen-Induced Microglial Activation and Dopaminergic Neurodegeneration
- Neuroprotective Properties of Naloxone
- Neuroprotective Effect of Other Anti-Inflammatory Agents
- Conclusion
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters