


Abstract
Adefovir is a nucleotide analog with anti-human immunodeficiency virus (HIV) activity that has been extensively studied in clinical trials. While on prolonged anti-HIV therapy with adefovir, some patients may develop drug-associated nephrotoxicity manifested by changes in laboratory markers of renal tubular functions that are reversible upon drug discontinuation. It has been recently shown that adefovir is efficiently transported by the human renal organic anion transporter 1 (hOAT1), a membrane transport protein localized in the kidney, that presumably mediates the accumulation of adefovir in renal proximal tubules. In an effort to look for novel inhibitors of this transport process, we used a cell line stably expressing hOAT1 to demonstrate that nonsteroidal anti-inflammatory drugs (NSAIDs) efficiently inhibit hOAT1-specific transport of adefovir at clinically relevant concentrations. Diflunisal, ketoprofen, flurbiprofen, indomethacin, naproxen, and ibuprofen were equally or more effective (IC50 = 0.85–8 μM) than probenecid or betamipron, two known potent inhibitors of hOAT1 (IC50 = 8 and 6 μM, respectively) with in vivo nephroprotective effects. Importantly, NSAIDs significantly reduced the shift in adefovir cytotoxicity observed upon hOAT1 expression with ketoprofen and naproxen being 2- to 3-times more effective than probenecid. Transport experiments with [3H]ketoprofen and [3H]ibuprofen revealed that NSAIDs themselves were not efficiently transported by hOAT1. None of the NSAIDs tested showed any interference with the anti-HIV activity of adefovir. In conclusion, these observations suggest that NSAIDs may reduce or delay the emergence of adefovir nephrotoxicity.
Renal organic anion transporter 1 (OAT1) localized in the basolateral membrane of renal convoluted tubules (Hosoyamada et al., 1999; Tojo et al., 1999) is an essential component of the tubular secretion of small negatively charged molecules (Sweet and Pritchard, 1999). In concert with the tubular brush-border membrane carriers/channels, OAT mediates active transepithelial movement of organic anions from systemic circulation into the glomerular filtrate (Pritchard and Miller, 1996). In addition to p-aminohippuric acid (PAH), a prototypical organic anion substrate, renal OATs from different mammalian species have shown broad substrate specificity with the capability of transporting endogenous metabolites (urate), signal molecules (cyclic nucleotides, prostaglandins) (Sekine et al., 1997), toxins (ochratoxin A) (Tsuda et al., 1999), xenobiotics (2,4-dichlorophenoxyacetic acid) (Villalobos et al., 1999), and fluorescent dyes (6-carboxyfluorescein) (Cihlar and Ho, 2000). Several recent studies indicated that the OAT system also is able to interact with a spectrum of important therapeutics, including β-lactam antibiotics (Jariyawat et al., 1999;Lin et al., 1999), and nonpeptidic angiotensin receptor antagonists (e.g., losartan and eprosartan) (Edwards et al., 1999), as well as nucleotide antiviral agents such as adefovir (Cihlar et al., 1999).
Adefovir [9-(2-phosphonomethoxyethyl)adenine] belongs to a class of acyclic nucleoside phosphonate analogs and exhibits a potent in vitro antiviral activity against herpes viruses and retroviruses (Cihlar and Bischofberger, 1998). Clinical trials in HIV-infected patients demonstrated antiviral efficacy of its orally available prodrug adefovir dipivoxil (120 mg once daily), against the wild type as well as various mutant drug-resistant HIV strains (Kahn et al., 1999). In addition, adefovir shows even more potent antiviral activity against hepatitis B virus in vitro, and several clinical studies are currently in progress to assess the utility of lower doses of adefovir dipivoxil (5–30 mg once daily) as a treatment for chronic hepatitis B virus infections.
During prolonged anti-HIV therapy with adefovir dipivoxil, some patients develop nephrotoxicity manifested as changes in laboratory markers of renal tubular functions, which are reversible upon treatment discontinuation (Kahn et al., 1999). Adefovir undergoes active tubular secretion (Cundy et al., 1995), indicating that its accumulation in proximal tubular cells may play a role in the etiology of this main dose-limiting adverse event. Our recent studies have shown that adefovir and cidofovir, a structurally related antiviral agent, are efficiently transported by the human renal organic anion transporter 1 (hOAT1) (Cihlar et al., 1999). Subsequent experiments have demonstrated a marked increase in adefovir and cidofovir cytotoxicity upon stable expression of hOAT1 in various types of mammalian cells, indicating that hOAT1 is directly involved in the mechanism of nephrotoxicity associated with these two antivirals (Ho et al., 2000). Notably, the specific increase in adefovir and cidofovir cytotoxicity due to hOAT1 expression was significantly reduced in the presence of probenecid, a potent inhibitor of hOAT1 transport activity. Although probenecid has shown in vivo nephroprotective effects (Lacy et al., 1998) and is currently coadministered together with cidofovir to reduce the potential for nephrotoxicity, its use as a nephroprotectant requires administration of a relatively high dose, which is often associated with gastrointestinal intolerance and other adverse effects.
Recently, a number of nonsteroidal anti-inflammatory drugs (NSAIDs) have been characterized for their interaction with rat OAT expressed inXenopus laevis oocytes. Several of the NSAIDs studied have shown potent inhibition of PAH uptake (Apiwattanakul et al., 1999). Therefore, in a search for novel inhibitors of hOAT1-mediated adefovir transport that could potentially serve as improved nephroprotective agents, we investigated the class of NSAIDs. Compared with probenecid, several NSAIDs showed superior efficacy with respect to the inhibition of hOAT1-specific adefovir transport as well as reduction of adefovir cytotoxicity mediated by hOAT1. Results from this study suggest that certain NSAIDs may reduce or delay the emergence of nephrotoxicity associated with adefovir and/or other drugs that accumulate in proximal tubules primarily via hOAT1.
Experimental Procedures
Materials.
[3H]Adefovir (30 Ci/mmol) was purchased from Moravek Biochemicals (Brea, CA). [3H]Ibuprofen (0.5 Ci/mmol) and [3H]ketoprofen (5 Ci/mmol) were obtained from American Radiolabeled Chemicals (St. Louis, MO). Nonradioactive adefovir was synthesized at Gilead Sciences as previously described (Holy and Rosenberg, 1987). Probenecid, betamipron (N-benzoyl-β-alanine), and all NSAIDs were from Sigma (St. Louis, MO).
Cells.
Generation and characterization of Chinese hamster ovary (CHO) cells stably expressing hOAT1 (CHOhOAT cells) and the control cells stably transfected with the empty pIRESneo expression vector (CHOpIRES cells) have been described previously (Ho et al., 2000). The cells were grown in phenol red-free F-12 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 mg/ml G-418. Cells for immediate use in the transport or cytotoxicity experiments were grown in the absence of antibiotics.
Transport Assays.
The assays were carried out in 12-well plates with nearly confluent cells as described previously (Ho et al., 2000). Briefly, the uptake of radiolabeled substrates in the presence or absence of varying inhibitor concentrations was determined at 37°C in Waymouth buffer. At the end of incubation, the cells were washed three times with ice-cold PBS and lysed directly on the plate in the presence of 0.5 ml/well 0.3% Triton X-100 for 15 min. Subsequently, the wells were washed with an additional 0.5 ml of detergent, the lysate and wash were combined, and the radioactivity in each sample was determined. Inhibitor concentrations reducing adefovir transport by 50% (IC50) were estimated from semilogarithmic plots of concentration versus percentage of uptake relative to uninhibited control.
Drug Cytotoxicity Assays.
CHOpIRES and CHOhOAT cells were seeded into 96-well plates at a density of 3 × 103 cells/well. After 24 h, various concentrations of the tested drugs were added in triplicate, and the cells were incubated for an additional 120 h. At the end of the incubation, cell viability was determined by a modified colorimetric assay with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (Ho et al., 2000). Average absorbance at 560 nm was calculated from triplicates, and the 50% cytotoxic concentration of each drug was determined from a semilogarithmic plot of drug concentration versus percentage of absorbance relative to untreated control.
Antiviral Assay.
MT2 T cells (gift from Dr. Norman Salzman, National Cancer Institute, Bethesda, MD) grown in RPMI-1640 medium supplemented with 5% fetal bovine serum, and antibiotics were infected with wild-type HIV-1 strain IIIB (Advanced Biotechnologies, Columbia, MD) at a multiplicity of infection equal to 0.01. The infected cells were distributed into a 96-well plate and various concentrations of adefovir in the absence or presence of various NSAIDs were added in triplicate. After 5 days, the virus-induced cell death was determined by using a colorimetric assay based on 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide as described (Weislow et al., 1989), and adefovir IC50 values were calculated.
Results
NSAIDs Are Potent Inhibitors of hOAT1-Mediated Uptake of Adefovir.
A number of structurally diverse NSAIDs were compared in CHOhOAT cells with respect to their inhibitory effect on the hOAT1-mediated transport of adefovir. All NSAIDs tested were able to reduce the accumulation of 10 μM [3H]adefovir in CHOhOATcells (Fig. 1). However, significant differences in their inhibitory potency were found. Diflunisal was the most potent inhibitor of adefovir uptake with an IC50 value of 0.85 μM. Ketoprofen, flurbiprofen, indomethacin, diclofenac, naproxen, and ibuprofen, with IC50 values ranging from 1.2 to 8 μM, were all equally or more effective than either probenecid or betamipron (IC50 = 8 and 6 μM, respectively), two known potent inhibitors of hOAT1. In contrast, etodolac, phenacetin, and piroxicam exhibited somewhat less inhibitory effect with IC50 values of 20 to 200 μM.

NSAIDs are potent inhibitors of hOAT1-mediated transport of adefovir. CHOhOAT cells were incubated with 10 μM [3H]adefovir and various concentrations of each inhibitor for 20 min at 37°C. IC50 values (mean ± S.D.; n= 2) were determined as described under Experimental Procedures.
hOAT1-Mediated Cytotoxicity of Adefovir Is Efficiently Reduced by NSAIDs.
Adefovir cytotoxicity increased significantly upon expression of hOAT1 in CHO cells (Fig.2). However, as shown previously, 1 mM probenecid efficiently reduced this hOAT1-mediated shift in the cytotoxicity of adefovir (Ho et al., 2000). Results demonstrating the inhibition of hOAT1-specific adefovir transport by NSAIDs suggested that this class of drugs might exhibit a protective effect similar to that of probenecid. Therefore, adefovir cytotoxicity was determined simultaneously in CHOhOAT and CHOpIRES cells in the presence or absence of 100 μM NSAIDs, a concentration at which the majority of NSAIDs did not produce any significant cytotoxic effects in the two cell lines (see below). Table 1 shows that a majority of the tested NSAIDs indeed decreased the cytotoxicity of adefovir in CHOhOAT cells. Among them, ketoprofen and naproxen were the most protective, reducing the cytotoxicity of adefovir 16- and 10-fold, respectively. In comparison, 100 μM probenecid showed less protective effect with adefovir cytotoxicity in CHOhOAT cells reduced approximately 5-fold. Ibuprofen, flurbiprofen, and diclofenac exhibited equal or slightly better protective effect than probenecid. As expected, none of the NSAIDs reduced cytotoxicity of adefovir in control CHOpIRES lacking hOAT1 transport activity. In these cells, a slight increase (2- to 4-fold) of adefovir cytotoxicity was observed in the presence of some NSAIDs. This could be due to the inhibitory effect that the drugs may exhibit against the cellular efflux of adefovir, similar to what has been shown previously with high concentrations of probenecid (Ho et al., 2000). To take this into account, the ratio between the adefovir 50% cytotoxic concentration in CHOhOAT and CHOpIRES cells was determined. The values revealed that in the presence of 100 μM ketoprofen and naproxen, the cytotoxicity of adefovir in CHOhOAT cells was only 7- and 12-fold higher, respectively, than that in CHOpIRES cells (Table1). In comparison, approximately 400-fold difference in adefovir cytotoxicity was observed between the two cell lines in the absence of hOAT1 inhibitors.
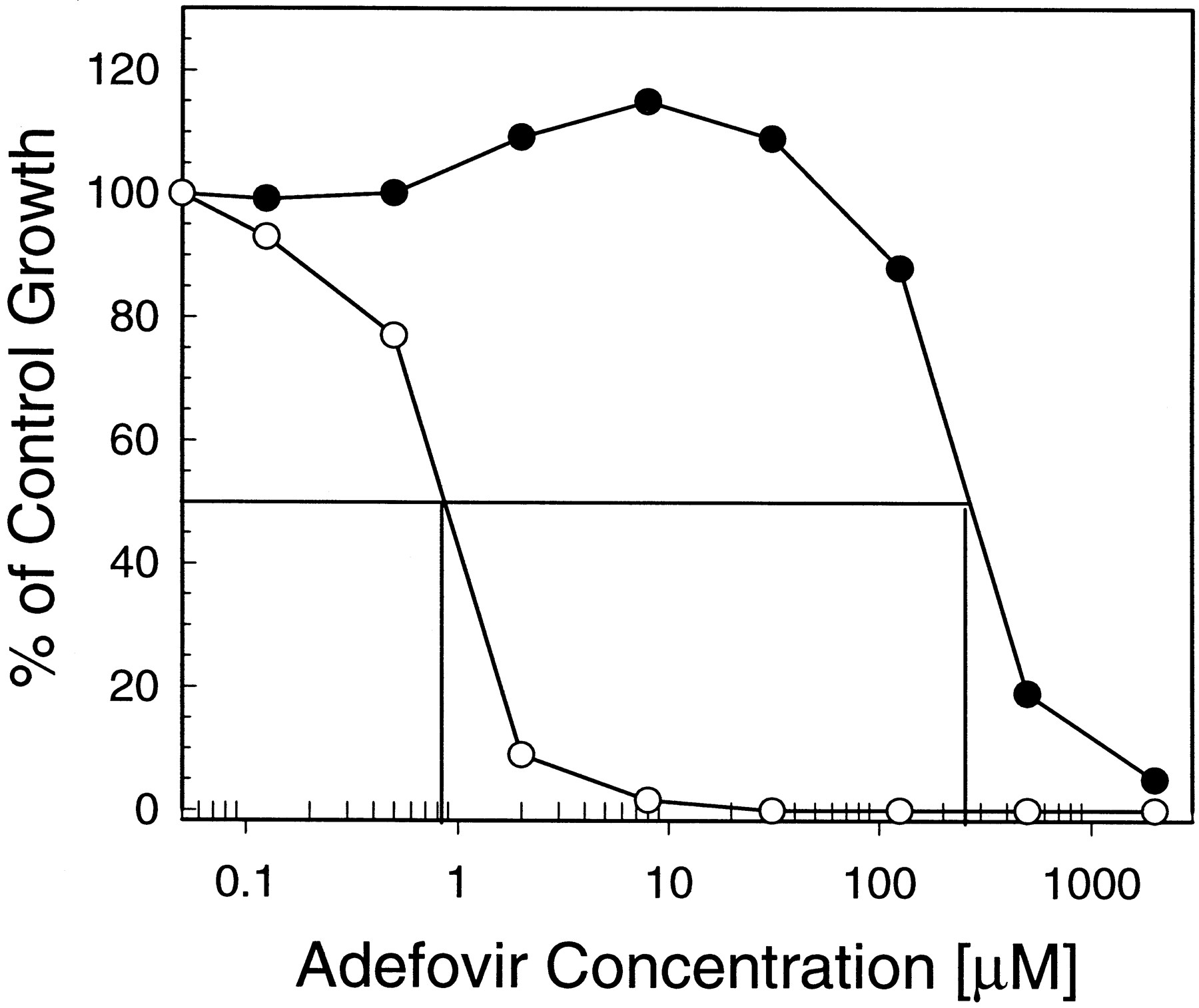
Effect of hOAT1 expression on the cytotoxicity of adefovir. Control cells (CHOpIRES; ●) and hOAT1-expressing cells (CHOhOAT; ○) in 96-well plates were incubated for 5 days with various concentrations of adefovir. Data are from a representative 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide-based cytotoxicity experiment performed in triplicate simultaneously with CHOpIRES and CHOhOATcells.
Effect of NSAIDs on hOAT1-mediated cytotoxicity of adefovir
NSAIDs Themselves Are Not Effectively Transported by hOAT1.
To address whether NSAIDs, besides being potent inhibitors of hOAT1, are also efficient substrates for this renal transporter, we compared intracellular accumulation of [3H]ketoprofen and [3H]ibuprofen in CHOpIRES and CHOhOAT cells. As shown in Fig. 3, no appreciable difference between the control and hOAT1-expressing cells was detected. In contrast, adefovir, an efficient hOAT1 substrate (Cihlar et al., 1999), accumulated in CHOhOAT cells to a level >30-fold higher than in CHOpIRES cells. In addition to the uptake experiments, the cytotoxicity of NSAIDs was compared in CHOpIRES and CHOhOAT cells. Again, contrary to what was observed with adefovir, the results showed no change in the cytotoxicity of NSAIDs upon hOAT1 expression (Table2). Altogether, these results indicate that NSAIDs are not efficiently transported by hOAT1.

Ibuprofen and ketoprofen are not efficiently transported by hOAT1. CHOpIRES cells (▪) and CHOhOAT cell (■) cells were incubated with 10 μM [3H]adefovir, [3H]ibuprofen, or [3H]ketoprofen for 60 min at 37°C. The samples were processed as described under Experimental Procedures. The data represent a mean of duplicates in a representative experiment.
NSAIDs exhibit no selective toxicity toward the CHOhOAT cell line
Adefovir Maintains Its Antiviral Activity in the Presence of NSAIDs.
Anti-HIV activity of adefovir in the presence and absence of selected NSAIDs was determined by the 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide-based colorimetric assay. Data in Table 3show that ketoprofen, naproxen, ibuprofen, and diflunisal did not interfere with the antiviral activity of adefovir in MT2 T cells infected with HIV-1. In fact, the anti-HIV activity of adefovir improved 2- to 5-fold in the presence of NSAIDs, which could be further evidence that NSAIDs may exhibit an inhibitory effect on cellular efflux of adefovir.
NSAIDs do not interfere with the anti-HIV activity of adefovir
Discussion
It is widely accepted that hOAT1 is a key component of the renal tubular secretory pathway of organic anions. A broad spectrum of molecules interact with this transporter, including the antiviral agent adefovir, which undergoes hOAT1-mediated transport at an efficiency comparable with that of PAH, a prototype hOAT1 substrate (Ho et al., 2000). As a consequence and possibly also due to the less efficient efflux across the tubular apical membrane, adefovir presumably accumulates within proximal tubules, as shown previously with the related antiviral nucleotide cidofovir (Cundy et al., 1996). In a proportion of HIV-infected patients treated with a long-term antiretroviral adefovir therapy, this may lead to a renal tubular dysfunction (Kahn et al., 1999).
In addition to hOAT1, expression of other membrane proteins capable of organic anion transport has been detected in kidney. They include OAT-K1 (Masuda et al., 1997), OAT-K2 (Masuda et al., 1999), OATP (Kullak-Ublick et al., 1995), oatp-2 (Noe et al., 1997), OAT2 (formerly known as NLT) (Sekine et al., 1998), OAT3 (Kusuhara et al., 1999), and OAT4 (Cha et al., 2000). Although some of these transporters may potentially contribute to a tubular accumulation of adefovir, several observations indicate that hOAT1 plays a crucial role in the etiology of adefovir nephrotoxicity. First, hOAT1-mediated transport of adefovir is highly efficient, and the heterologous expression of hOAT1 in various mammalian cells induces cytotoxicity of adefovir (Ho et al., 2000). Second, hOAT1 is the only organic anion transporter shown to be expressed at high level specifically in the kidney (Cihlar et al., 1999). This corresponds with the organ-specific toxicity of adefovir. Third, localization of hOAT1 to the basolateral membrane of proximal tubules (Hosoyamada et al., 1999; Tojo et al., 1999) is consistent with the changes in specific markers for proximal tubular functions associated with adefovir nephrotoxicity (Kahn et al., 1999). Fourth, in vivo nephrotoxicity potential of the closely related nucleotide cidofovir and its prodrug correlates with the efficiency of their hOAT1-mediated transport (Ho et al., 2000). Altogether, these observations suggest that nephrotoxicity of adefovir might be reduced by coadministration of a potent hOAT1 inhibitor, as demonstrated previously for the nephrotoxicity associated with cidofovir and β-lactam antibiotics where probenecid and betamipron, respectively, served as nephroprotectants (Lacy et al., 1998; Kim et al., 1999).
To search for novel hOAT1 inhibitors, we have recently established an in vitro model based on CHO cells stably expressing hOAT1 (Ho et al., 2000). In this model, we demonstrated that a number of NSAIDs could inhibit adefovir transport via hOAT1. Previous studies revealed that a “prototypic” substrate and/or inhibitor of renal OAT1 system consists of a hydrophobic core to which at least one negatively charged group, often a carboxyl, is attached (Ullrich, 1997). The NSAIDs selected for this study varied structurally both in the hydrophobic core and in the carboxylic acid moiety. In general, the inhibitory potency did not seem to be significantly influenced by the type of carboxylic acid. Diflunisal, the most potent inhibitor of adefovir transport, which contains a carboxyl moiety attached directly to the hydrophobic aromatic ring, was only slightly more potent than some of the NSAIDs containing a propionic acid side chain (ketoprofen, flurbiprofen, naproxen) or acetic acid side chain (indomethacin, diclofenac). However, etodolac, which is also a derivative of acetic acid, was a poor hOAT1 inhibitor. Etodalac was the only NSAID with an alkyl substituent in the immediate vicinity of the carboxylic acid side chain, which presumably decreases the planarity of the molecule and may reduce its interaction with the substrate-binding site of hOAT1. Two NSAIDs lacking the carboxyl moiety (phenacetin and piroxicam) clearly showed less inhibitory effect. With respect to the hydrophobic part of the molecule, two aromatic rings linked by a short spacer appear to be characteristic for NSAIDs with the highest inhibitory potency (diflunisal, ketoprofen, and flurbiprofen). Similar structural features have been found for NSAIDs interacting strongly with the rat homolog of hOAT1 (Apiwattanakul et al., 1999). Although PAH was used in that study as a substrate for the rat transporter, naproxen, indomethacin, piroxicam, and phenacetin exhibited a relative inhibitory potency almost identical with that observed with hOAT1 and adefovir, indicating a significant similarity between the two transport proteins. Rat OAT1 indeed shares 88% amino acid residues with hOAT1 and shows a higher degree of homology with hOAT1 than any of the other animal OATs identified thus far (Cihlar et al., 1999).
As a consequence of a markedly higher intracellular accumulation of adefovir and its metabolites, the expression of hOAT1 increases cytotoxicity of adefovir in CHO cells by 400-fold. However, in the presence of 1 mM probenecid, this susceptibility shift can be reduced approximately 50-fold (Ho et al., 2000). Thus, it appears that the cytoprotective effect of probenecid is proportional to its concentration because at 100 μM, it reduces the hOAT1-mediated cytotoxicity of adefovir only 5-fold. Comparison with probenecid showed that at least two of the NSAIDs were more cytoprotective than probenecid; several others exhibited comparable effects. In general, there was a correlation between the inhibitory potency of NSAIDs in the adefovir transport assay and their cytoprotective effect. Ketoprofen, flurbiprofen, and naproxen were potent inhibitors of adefovir transport and exhibited cytoprotection superior to that of probenecid. For diclofenac and ibuprofen, both the inhibitory activities and the cytoprotective effects were comparable with those of probenecid. Diclofenac, the most potent hOAT1 inhibitor among the tested NSAIDs, still showed a distinct cytoprotective effect, despite being tested at a significantly lower concentration due to its intrinsic cytotoxicity. Furthermore, piroxicam, one of the least potent inhibitors, showed only a minor reduction in the hOAT1-specific cytotoxicity of adefovir. In contrast, indomethacin did not exhibit almost any cytoprotection in hOAT1-expressing cells despite its efficient inhibition of hOAT1 transport activity. This discrepancy may be, at least in part, explained by a relatively high nonspecific cytotoxicity of indomethacin.
Although potent inhibitors of hOAT1, neither ketoprofen nor ibuprofen was an efficient substrate. In correlation, the expression of hOAT1 did not induce any changes in the cytotoxicity of ketoprofen and ibuprofen or the other tested NSAIDs. Studies with rat OAT1 have demonstrated a competitive type of inhibition (Apiwattanakul et al., 1999), indicating that NSAIDs can presumably bind into the transporter active site, but the transporter is not capable of their efficient translocation across the plasma membrane. Despite the lack of transport via hOAT1, previous studies have indicated an active accumulation of some NSAIDs in rat renal proximal tubular cells (De Zeeuw et al., 1988). Thus, it is possible that the tubular uptake of NSAIDs may be mediated by some of the other renal organic anion transporters. Indomethacin and ketoprofen interact with OAT-K1 and OAT-K2, two recently identified transporters expressed in kidney (Masuda et al., 1997, 1999). In addition, some of the other above-mentioned renal organic anion transporters may contribute to the tubular transport of NSAIDs.
Given the lack of hOAT1 expression in T cells (Cihlar et al., 1999) and the nonspecific fluid-phase endocytosis being the proposed mechanism of adefovir uptake into T cells (Olsanska et al., 1997), one would not expect any compromising effect of NSAIDs on the anti-HIV activity of adefovir. Indeed, none of the NSAIDs that showed the most potent inhibition of hOAT1 interfered with the antiviral activity of adefovir when tested in HIV-1-infected MT2 cells. Interestingly, the antiviral effect of adefovir improved 2- to 5-fold in the presence of the tested NSAIDs. This could possibly be due to their specific effect on adefovir efflux from the host cells, which is presumably actively mediated by the efflux pump multidrug resistance protein 4 (Schuetz et al., 1999). Similarly, an enhancement of the in vitro cytotoxicity of various anticancer drugs in the presence of NSAIDs has been previously observed in specific tumor cell lines. Indomethacin, sulindac, and some other NSAIDs have been shown to potentiate the cytotoxicity of doxorubicin, daunorubicin, teniposide, and vincristine, and to inhibit the cellular efflux of specific multidrug resistance protein substrates (Duffy et al., 1998).
In conclusion, our study indicates that various NSAIDs are equally or more potent inhibitors of hOAT1 than probenecid and exhibit protective effects against hOAT1-mediated cytotoxicity of adefovir. Given the known effect of probenecid as an in vivo nephroprotectant, some of the NSAIDs also may reduce the nephrotoxic potential of certain therapeutics such as adefovir, assuming that these are accumulated in proximal tubules primarily via hOAT1. Importantly, the inhibitory potency of ketoprofen, ibuprofen, naproxen, and other NSAIDs is within the range of their clinically relevant plasma concentrations (Ishizaki et al., 1980; Albert and Gernaat, 1984), indicating that they may indeed exhibit in vivo nephroprotective effects when coadministered with nephrotoxic hOAT1 substrates. It should be noted, however, that chronic therapy with high doses of certain NSAIDs might, in some cases, be associated with renal insufficiency, papillary necrosis, and other renal effects, especially in elderly patients and patients with impaired renal function (Stillman, 1989; Bennett et al., 1996). In addition, hepatotoxicity associated with certain NSAIDs has been reported (Rabinovitz and Van Thiel, 1992; Bjorkman, 1998). Although the incidence of these events is relatively low (Bjorkman, 1998; Whelton, 1999), further in vivo studies would be needed to assess the proper therapeutic dose of NSAIDs when used as nephroprotectants.
Acknowledgments
We thank Craig Gibbs, Bill Lee, Norbert Bischofberger, and Mick Hitchcock of Gilead Sciences for critical reading of the manuscript and for valuable comments and suggestions.
Footnotes
-
Send reprint requests to: Tomas Cihlar, Gilead Sciences, 333 Lakeside Dr., Foster City, CA 94404. E-mail:tomas_cihlar{at}gilead.com
- Abbreviations:
- OAT1
- organic anion transporter 1
- PAH
- p-aminohippuric acid
- hOAT1
- human renal organic anion transporter 1
- NSAIDs
- nonsteroidal anti-inflammatory drugs
- CHO
- Chinese hamster ovary
- CHOhOAT
- CHO cells stably expressing hOAT1
- CHOpIRES
- CHO cells stably transfected with the expression vector pIRES-neo
- Received March 14, 2000.
- Accepted May 31, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}