


Abstract
Life-threatening diarrhea afflicts a considerable percentage of patients treated with irinotecan, an anticancer agent with effects elicited through its active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). The primary detoxification pathway for SN-38 is glucuronidation. The purpose of this study was to evaluate the role that intestinal UDP-glucuronosyltransferases (UGTs) have from hepatic UGTs in modulating this diarrhea. To investigate this, Gunn rats devoid of UGT1A activity were injected with recombinant adenoviral vectors expressing UGT1A1, 1A6, and 1A7, resulting in reconstituted hepatic UGT expression comparable to a heterozygote. Hepatic microsome studies indicated that 4 to 7 days after adenoviral injection, transfected Gunn rats (j/jAV) had SN-38 glucuronide (SN-38G) formation rates three times higher than control heterozygote rats (j+AV). The adenovirus did not impart any glucuronidating capacity to the intestine in j/jAV rats, whereas j+AV rats possessed intestinal UGT function. After the administration of 20 mg/kg/day irinotecan i.p. to j/jAV rats 4 days after adenovirus injection, diarrhea ensued before the fourth irinotecan dose. j+AV rats were spared the diarrhea, and the toxicity was mild compared with the j/jAV rats, as measured by diarrhea scores, weight loss, and histological assessments of the cecum and colon. The pharmacokinetics of irinotecan, SN-38, and SN-38G indicate that the systemic exposure of SN-38 and SN-38G was higher and lower, respectively, in j/jAV rats. Despite this, the biliary excretion of irinotecan and metabolites was similar. Because intestinal UGTs are the main discriminating factor between j/jAV and j+AV rats, their presence seems to be critical for the gastrointestinal protection observed in j+AV rats.
The UDP-glucuronosyltransferases (UGTs) are a superfamily of metabolic enzymes that catalyze the transfer of UDP-glucuronic acid to endogenous and xenobiotic substrates. Glucuronidation is a detoxification mechanism from several standpoints. It decreases the apparent volume of distribution, increases the molecular weight, and increases substrate specificity for active transport by imparting a negative charge, which are all processes that facilitate substrate elimination from the body (Guillemette, 2003). Furthermore, in most cases, glucuronidation renders the substrate inactive with respect to its pharmacological or physiological target. A Wistar-derived rat model of UGT1A subfamily deficiency, the Gunn rat, has allowed tremendous insight into the importance of the UGT1A family in the metabolism and toxicity of substrates (Wells et al., 2004). The abolition of UGT1A glucuronidation in these rats stems from a frameshift mutation that yields a truncated, nonfunctional protein unable to bind UDP-glucuronic acid (Iyanagi et al., 1989).
Although the primary organ of glucuronidation receiving most attention has been the liver, research on intestinal UGTs has shown their importance. Large differences in protein levels of the UGT1A family are not observed between these organs, and hepatic and intestinal tissues have been shown to have similar catalytic rates for a number of substrates, including carcinogens, tertiary amines, and steroids (Strassburg et al., 1999, 2000). Differences in the tissue glucuronidation profile for a given substrate may be due to tissue-specific UGT isoform expression, evident in both humans and rodents (Strassburg et al., 1999; Shelby et al., 2003; Gregory et al., 2004). Because of their localization, intestinal UGTs have been shown to contribute significantly to first-pass metabolism after oral administration of substrates, and they may limit the systemic exposure of substrates undergoing enterohepatic recirculation (Sfakianos et al., 1997; Chen et al., 2003).
SN-38, the active metabolite of the chemotherapeutic drug irinotecan (Camptosar, Pfizer, Inc., New York, NY), is a substrate for hepatic and intestinal UGT1A isoforms in both rats and humans (Tallman et al., 2005). SN-38 glucuronide (SN-38G), the resulting metabolite, is inactive and nontoxic (Fig. 1). By injuring enteric cells, SN-38 causes a dose-limiting diarrhea in approximately 20% of patients (Araki et al., 1993; Iyer et al., 1998; Saltz et al., 2000). Several strategies have been used in research studies to prevent this diarrhea. These have focused mostly upon reducing intestinal SN-38 concentrations and include the inhibition of SN-38 and SN-38G biliary excretion and the inhibition of β-glucuronidase, an intestinal enzyme that will hydrolyze SN-38G to form SN-38 (Takasuna et al., 1996; Kehrer et al., 2001; Horikawa et al., 2002; Desai et al., 2005). Likewise, it was proposed by Gupta et al. (1994) that patients with poor hepatic SN-38 glucuronidation would have high biliary SN-38 concentrations and probably experience gastrointestinal toxicity. They devised a mathematical equation to describe this scenario, given as the ratio of the area under the curve (AUC) values of SN-38 to SN-38G multiplied by irinotecan AUC. In this study, the values, termed “biliary index”, were significantly higher in patients with diarrhea (Gupta et al., 1994). However, the biliary index has not been reproducibly validated by other investigators (Canal et al., 1996; Xie et al., 2002), probably because SN-38G excreted in the bile also contributes to toxicity after cleavage with β-glucuronidase by forming SN-38.

Glucuronidation of SN-38 after carboxylesterase cleavage of irinotecan. UGT1A1, -1A7, and -1A9 have been identified as the major isoforms responsible for SN-38 catalysis in humans.
Thus far, the approaches to both predict and prevent SN-38-mediated diarrhea have not explored the capacity for inherent enterocyte protection through intestinal UGT expression. This concept of intestinal UGT protection in irinotecan gastrointestinal toxicity was proposed by Tukey et al. (2002); yet, thus far, it has not been investigated, probably because of the difficulty in isolating their contribution from hepatic UGTs in a toxicity model that takes several days to develop (Tukey et al., 2002). To address the hypothesis that intestinal UGTs play a major role in protecting against SN-38-induced diarrhea, we used a Gunn rat model with reconstituted hepatic UGT activity, achieved by adenoviral gene delivery. Intravenous injection of adenoviral vectors carrying rat UGTs will exclusively infect the liver (Amalfitano, 2004), and in specific doses of UGT1A1, -1A6, and -1A7, they have been shown to elicit a hepatic glucuronidation pattern similar to a Wistar/Gunn heterozygote (Miles et al., 2006). By comparing the pharmacokinetic and toxicity profiles from these transfected Gunn rats, possessing only hepatic UGTs, to those of heterozygous Gunn rats, possessing both hepatic and intestinal UGTs, we show that intestinal conjugation of SN-38 is critical in reducing the incidence of gastrointestinal toxicity.
Materials and Methods
Materials. Irinotecan and SN-38G methyl ester were kindly provided by Dr. Robert Kelly (Pfizer, Inc., Kalamazoo, MI). Preparation and quantitation of SN-38 and SN-38G were described previously (Tallman et al., 2005). Reagents for microsomal experiments were purchased through Sigma-Aldrich (St. Louis, MO). Electrophoresis, gel/membrane transfer boxes, and Western blot reagents were obtained from Bio-Rad (Hercules, CA), unless stated otherwise. All other chemicals and solvents were purchased through commercial sources.
Generation and Propagation of Recombinant Adenoviruses. First generation adenoviral vectors carrying rat UGT1A1, -1A6, -1A7, or -1A10 isoforms were produced using the homologous recombination method in Escherichia coli by the Massey Cancer Center Virus Vector Shared Resource Facility at Virginia Commonwealth University. The vector plasmid pTG-cytomegalovirus contained the E1- and E3-deleted adenoviral strain H5d1324. The shuttle plasmid pZero-TG-cytomegalovirus-rUGT1A contained a cloned rat UGT isoform within the E1-deleted region. After the vector plasmid was cut with restriction enzyme ClaI, both plasmids were cotransformed into E. coli. Selected recombinant plasmids were transfected into human embryonic kidney 293B cells, which provide the deleted viral E1 proteins necessary for reproduction, and they were used to propagate the viruses (American Type Culture Collection, Manassas, VA). Replication-deficient viruses were then isolated from cellular components through freeze-thaw and centrifugation cycles, and finally they were purified and concentrated through cesium chloride gradients and dialysis (Miles et al., 2006). By lysing and measuring the absorbance of an aliquot of diluted virus at 260 nm, the final viral concentration was determined and given in optical density (OD) units, where 1 OD260 is 1 × 1012 viral particles per milliliter of dialysate (Herrmann et al., 2004). Adenoviral stocks were stored at –80°C.
Animals. Male Gunn (j/j) and heterozygous (j+) Gunn rats (weighing 200–275 g) used in this study were colony-bred from j/j males and j+ females at Virginia Commonwealth University (Richmond, VA). Gunn rats were selected from j+ littermates by jaundice evident on their feet and ears. During the study, animals were housed two per cage in a temperature- and humidity-regulated room with a 12-h light/dark cycle. Standard rat chow and water were made freely available. All care and conducted procedures were approved by the University of North Carolina (Chapel Hill, NC) and Virginia Commonwealth University, Institutional Animal Care and Use Committees.
Animal Treatment and Toxicity. On day 1, j/j rats (n = 5) received 0.15 ml of adenovirus as a bolus through the tail vein. The total dose of 0.13 OD of adenovirus consisted of 0.1 OD of adenovirus encoding rat UGT1A1, 0.025 OD of UGT1A6, and 0.005 OD of UGT1A7. To control for any effects adenoviral injection may have in the j/j rats, j+ rats (n = 5) received 0.13 OD units of adenovirus carrying rat UGT1A10, an isoform with no activity toward SN-38 (data not shown). Hepatic UGT expression in the infected Gunn (j/jAV) animals was deemed adequate 4 days after adenoviral infection, as determined by visually inspecting the plasma for the resolution of hyperbilirubinemia. Thus, 4 days after infection, j/jAV and infected heterozygote Gunn (j+AV) rats received 20 mg/kg of commercially available irinotecan (20 mg/ml Camptosar; Pfizer, Inc.) in a final volume of 0.4 ml in their intraperitoneal space. Blood (0.1 ml) was then taken at 0.25, 0.5, 1, 2, 4, 8, and 12 h after this first dose of irinotecan. The blood was centrifuged, and plasma was stored at –20°C until analysis. Irinotecan was administered daily to j/jAV and j+AV for 3 days. Before each dose of irinotecan, the rats were weighed and assessed for the extent of diarrhea on a scoring system: 0, firm stool; 1, malformed stool; 2, watery stool with perianal staining; and 3, severe perianal staining (Takasuna et al., 1996). On the seventh day after adenovirus administration, the animals were euthanized. Livers from each rat were placed at –80°C. Colons and cecums were rinsed and placed in 10% formalin (Fisher Scientific, Hampton, NH). Pilot studies using irinotecan doses up to 80 mg/kg determined the dosing schedule used in the larger study described previously. Irinotecan 20 mg/kg × three doses was chosen because diarrhea was observed with this regimen, yet it provided the lowest cumulative dose of irinotecan given.
In a separate study, j/jAV (n = 4) and j+AV (n = 2) rats were anesthetized, and biliary catheters were placed 4 days after adenovirus injection with the isoforms and doses as described above for the respective genotype. Biliary cannulas were placed, and bile was collected under anesthesia over the following intervals: 0 to 0.25, 0.25 to 0.5, 0.5 to 1, 1 to 2, 2 to 4, and 4 to 6 h. After the final collection interval was completed, rats were euthanized. Livers from each rat were placed at –80°C.
Microsomal Preparation and Glucuronidation Assay. Livers from all rats in the biliary excretion study and the toxicity/pharmacokinetic study were used to make hepatic microsomes. Intestines and colons from irinotecan-naive j/jAV and j+AV (n = 2 in each group) rats were extracted 7 days after adenovirus injection and used to make microsomes. The procedures followed to make microsomes from these organs were described previously (Tallman et al., 2005). Microsomal protein concentrations were determined by the Bradford method, using bovine albumin as a standard.
In vitro conjugation reactions of SN-38 with the hepatic and intestinal microsomes contained the following components: magnesium chloride (10 mM assay concentration), Brij 35 (0.5 mg/mg protein), d-saccharic acid 1,4-lactone (10 mM), SN-38 carboxylate (300 μM), and microsomal protein (0.25 mg/ml) in a final volume of 200 μl of 0.1 M Tris, pH 7.0. The reaction was initiated by the addition of UDP-glucuronic acid (2 mM final concentration) and proceeded at 37°C for 40 min and quenched with acetonitrile.
Western Blotting. Hepatic, intestinal, and colonic microsomal proteins (50 μg) were subjected to electrophoresis through a 4 to 15% gradient Tris gel (Ready Gel; Bio-Rad). Upon separation, proteins were transferred to nitrocellulose membranes using 105 V for 1 h. Membranes were then washed in 5% milk/0.2% Tween in Tris-buffered saline (TBS-T) for 1 h to prevent nonspecific antigen-antibody binding. After two washes in 0.5% TBS-T, blots with hepatic microsomes were incubated with either α-rat UGT1A1 (diluted 1:1000 in 0.5% milk/0.2% TBS-T) or α-rat UGT1A7 (1:1500 in 0.5% milk/0.2% TBS-T) for 1 h each. Membranes with intestinal and colonic microsomes were probed with α-UGT1A antiserum (1:1000 in 0.5% milk/0.2% TBS-T) overnight. Information on specificity and production of antiserum reacting with rat UGT1A1, UGT1A7, or with the UGT1A common region on all rat UGT1A proteins was published previously (Kessler et al., 2002; Webb et al., 2005). Antiserum was washed off twice with 0.5% TBS-T, and a horseradish peroxidase-conjugated α-mouse IgG (1:10,000; Chemicon International, Temecula, CA) was incubated with all blots for 1 h. After two washes with 0.5% TBS-T, chemiluminescent reagent (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK) was applied to hepatic microsomal membranes, exposed to film (BioMax; Eastman Kodak, Rochester, NY) for 4 min, and developed. For intestinal protein blots, chemiluminescent reagent (SuperSignal; Pierce Chemical, Rockford, IL) was applied, and a PhosphorImager (Versadoc; Bio-Rad) was used to visualize the resulting bands. In all cases, a molecular mass marker was added to the gel to verify the presence of the UGT band (approximately 55 kDa).
Chromatographic Analysis. High-performance liquid chromatography (HPLC) with fluorescence detection was used to quantify irinotecan, SN-38, and SN-38G concentrations from plasma, bile, and in vitro microsomal incubations. Acetonitrile (300 μl), internal standard (camptothecin; 200 ng), and perchloric acid (5 μl; 5% solution) were added to each plasma sample (25 μl). Bile was first diluted 1 to 200 in Tris, and 200 μl was subject to acetonitrile (800 μl) and perchloric acid precipitation. Camptothecin (100 ng) was added to bile samples as an internal standard. For microsomal samples, acetonitrile (800 μl) containing perchloric acid (5%; 5 μl) and irinotecan as an internal standard (50 ng) were added. All samples were then centrifuged at 15,000g for 10 min. Next, the acetonitrile layer was extracted and evaporated under a stream of nitrogen. Samples were then reconstituted with 200 μl of 30% methanol/100 mM ammonium acetate. Standards were prepared as described above for each matrix and contained blank bile, blank plasma, or microsomal protein.
The HPLC system used to inject, separate, and detect analytes included an isocratic pump (LC-600; Shimazdu, Tokyo, Japan), autosampler (SIL10A; Shimadzu), fluorescence detector (HP1046A; Hewlett Packard, Palo Alto, CA), and a reversed-phase column (Hypersil BDS C-18, 5 μm, 150 × 4.6 mm]; Thermo Electron Corporation, Woodstock, GA). The HPLC method was adapted from a method published previously (Sparreboom et al., 1998). The mobile phase consisted of 37% methanol/100 mM ammonium acetate and 5 mM tetrabutylammonium sulfate, pH 4.9, run at 1.5 ml/min for microsomal samples. For plasma and bile samples, 39% methanol/100 mM ammonium acetate and 15 mM tetrabutylammonium sulfate, pH 4.9, was run as mobile phase at 1.3 ml/min. Standard curves in each matrix were linear and validated if the lower limit of quantitation coefficient of variance was ≤15% and the analyte-to-internal standard ratio normalized to concentration was constant. Calibration curve ranges were as follows: for plasma, 2.5 to 200 ng/ml SN-38G, 100 to 10,000 ng/ml irinotecan, and 2.5 to 50 ng/ml SN-38; for bile, 1.25 ng to 100 ng/ml SN-38G, 12.5 ng to 1000 ng/ml irinotecan, and 2.5 to 75 ng/ml SN-38; and for microsomes, 2 to 300 ng/ml SN-38G. Excitation and emission wavelengths were 229 and 420 nm, respectively, for SN-38G, irinotecan, and camptothecin. The emission wavelength used to detect SN-38 was changed to 543 nm during each chromatographic run. To avoid interday variability, all samples in a given matrix were processed and run on the same day. A representative chromatograph for bile, plasma, and microsomes is given in Fig. 2.
Histology. Colon and cecum samples were removed from formalin, rinsed, and stored in 70% ethanol. Samples were then embedded in paraffin, sectioned, mounted on slides, and stained with hematoxylin and eosin by the Histopathology Core Lab (Lineberger Comprehensive Center, University of North Carolina).
Data Analysis. Pharmacokinetic parameters for irinotecan, SN-38, and SN-38G were determined by noncompartmental analysis using WinNonlin software version 5.0 (Pharsight, Mountain View, CA). Statistical tests were performed using SigmaStat software version 2.0 (Systat Software, Inc., Point Richmond, CA). A t test was used to assess differences between j/jAV and j+AV rats given irinotecan, because the data were found to be normally distributed with equal variance. The Mann-Whitney rank sum test was used to analyze differences between the groups with respect to diarrhea scores. To assess biliary excretion, the amounts of irinotecan and metabolites excreted over the collection intervals were summed. Statistical significance was reached at p ≤ 0.05.

Chromatographs of SN-38G (1), irinotecan (2), camptothecin (3), and SN-38 (4) standards in bile, diluted 1:200, 200 μl (A); in plasma, 25 μl (B); or in microsomal reaction, 200 μl (C). Concentrations are 1, 25 ng/ml; 2, 250 ng/ml; and 4, 50 ng/ml (A); 1, 100 ng/ml; 2, 10 μg/ml; and 4, 100 ng/ml (B); and 1, 125 ng/ml (C).
Results
All rats injected with adenovirus tolerated the dose well, with no immunological response or behavioral or pathophysiological changes evident after administration. Gunn rats receiving 0.13 OD of adenovirus expressed rat UGT1A1 and UGT1A7, the isoforms relevant to SN-38 glucuronidation, in the liver (Fig. 3). By visual inspection of the blots and glucuronidation profiles in individual rats (n = 5/group), it seems that rat UGT1A7 expression is more highly associated with SN-38 glucuronidation compared with the expression of rat UGT1A1 (Fig. 3). In the j/jAV rats (n = 4), hepatic SN-38 glucuronidation reached its maximal expression 4 days after adenoviral infection (average of 38.8 pmol of SN-38G formed/min/mg protein), and it was sustained over the experimental period to 7 days postinfection (average of 34.8 SN-38G formed/min/mg protein). Over this time frame, the j/jAV rats have an average of three to four times greater SN-38 hepatic glucuronidation capacity than the j+AV rats (Figs. 3 and 4). The difference in activity between j/jAV and j+AV rats was significant at 7 days postinfection (p = 0.003) (Fig. 3). Hepatic glucuronidation of SN-38 in the j/jAV rats was solely due to the adenovirus delivery, because uninfected Gunn rats possess no intrinsic SN-38 catalytic activity (data not shown). Figure 5 reveals that an intravenous injection of adenovirus does not impart any UGT1A expression or SN-38-conjugating activity to the intestine or colon of a j/jAV rat (n = 2 rats/intestinal segment).
The pharmacokinetic profile and parameters of irinotecan and its metabolites are shown in Fig. 6 and Table 1. The pharmacokinetics of irinotecan were similar between j/jAV and j+AV rats (n = 5/group). SN-38 exposure, as represented by AUC, was significantly higher in j/jAV rats. The elimination half-life of SN-38G was not calculated due to the lack of a reproducible terminal concentration/time slope in the profiles of most rats. Despite higher in vitro SN-38 glucuronidation rates, the AUCSN-38G in j/jAV rats was approximately 2-fold lower than j+AV rats on average.
Pharmacokinetic parameters after a single dose of irinotecan (20 mg/kg i.p.) in j/jAV and j+AV rats (n = 5 each) as determined by noncompartmental analysis
AUC refers to that determined over the blood collection interval from 0 to 12 h. Values are given as mean ± S.E.

Hepatic expression of rat UGT1A1 (A) and rat UGT1A7 (B) in j+ AV rats (lanes 1–5) and j/jAV rats (lanes 6–10) 7 days after adenoviral injection of rat UGT1A10 to j+ rats or UGT1A1, -1A6, and -1A7 to j/j rats. Corresponding in vitro SN-38 glucuronidation rates for j+AV (open bars, 1–5) and j/jAV (closed bars, 6–10) (C). Bars represent data from individual rats and are an average of duplicate activity measurements. Hepatic microsomes were prepared from rats involved in the pharmacokinetic/toxicity study.
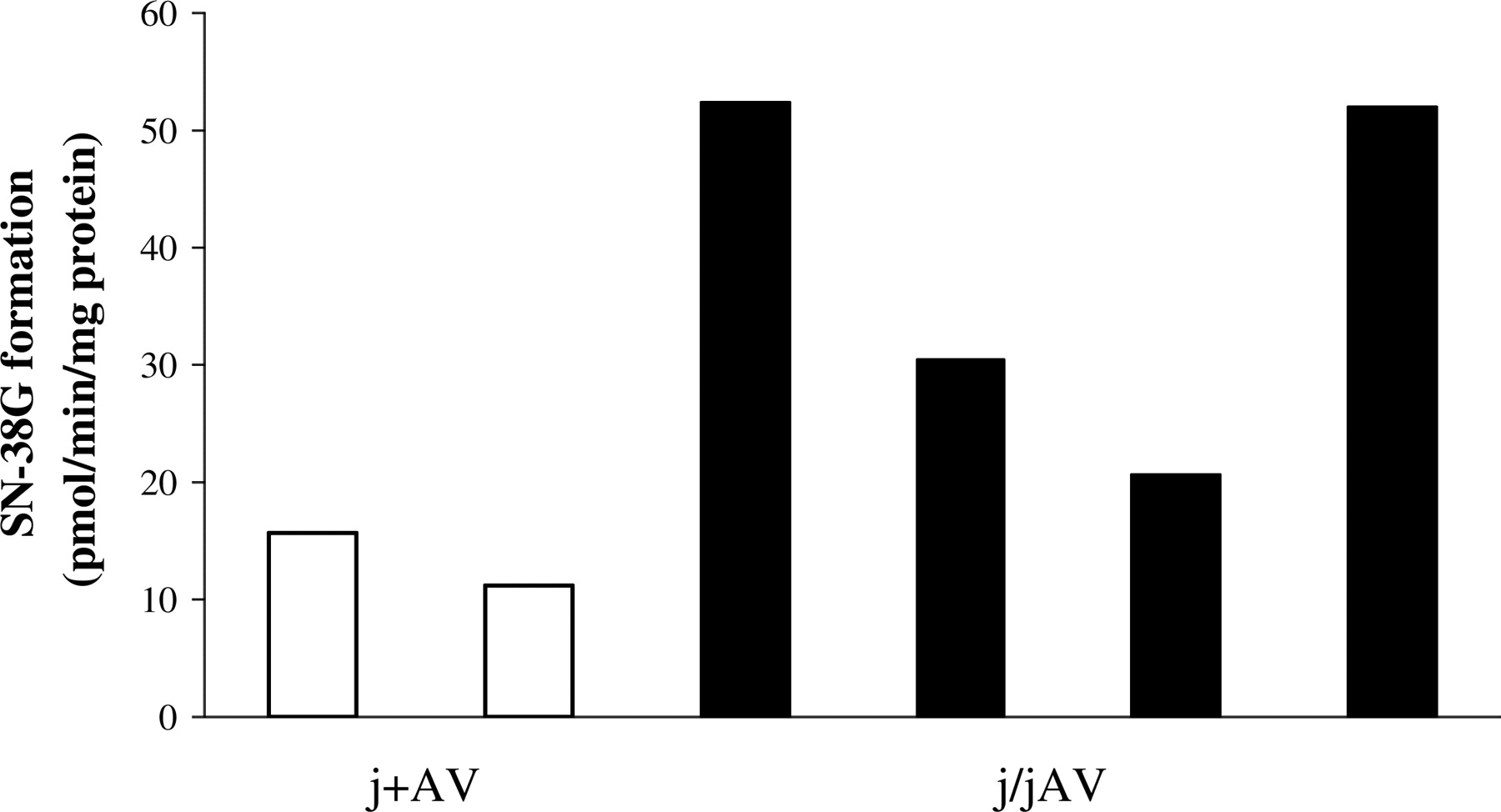
Hepatic SN-38 glucuronidation rates for j+AV (open bars) and j/jAV (closed bars) rats 4 days after adenoviral injection of 0.13 OD rat UGT1A10 or -1A1, -1A6, and -1A7, respectively. Bars represent data from individual rats and are an average of duplicate activity measurements. Hepatic microsomes were prepared from rats used for the biliary excretion study. Rats received one dose of irinotecan 20 mg/kg/day i.p.

Intestinal and colonic rat UGT1A expression (A) and SN-38 glucuronidation rates (B) for j/jAV and j+AV rats (n = 2 rats per group and intestinal segment). Open and filled bars represent SN-38 glucuronidation rates in the j+AV rats in the colon and intestine, respectively. Each bar denotes data from individual rats and is an average of duplicate activity measurements. Blots and in vitro activities were determined from intestinal and colonic microsomes of j+AV and j/jAV rats made 7 days after adenoviral injection of 0.13 OD rat UGT1A10 or -1A1, -1A6, and -1A7, respectively. Due to the poor condition of the gastrointestinal tract of j/jAV rats treated with irinotecan, all intestinal microsomes were processed from irinotecan-naive rats.
In a separate experiment to determine whether intestinal exposure via bile to irinotecan, SN-38, and SN-38G was similar between j/jAV (n = 4) and j+AV (n = 2) rats, bile was collected from rats from both groups after a single dose of irinotecan 20 mg/kg i.p. 4 days after adenovirus injection. Although statistical analysis is not applicable, it seems that irinotecan was excreted to a greater degree in j+AV rats relative to j/jAV rats over a 6-h period (Fig. 7). Little or no difference in the excretion of SN-38 and SN-38G between the two groups was apparent.
The two groups of rats responded to repeated doses of irinotecan with dissimilar results. Twenty-four hours after the second and third dose of irinotecan (20 mg/kg/day i.p.), j/jAV rats were found to have lost a greater percentage of their body weight relative to baseline (Fig. 8). After finding that several j/jAV rats had reached the predetermined maximum acceptable weight loss limit of 20% the morning after the third irinotecan dose was administered, the study was ended, and all animals were euthanized. At this time, every j/jAV rat (n = 5) had at least grade 2 diarrhea characterized by loose stools and perianal staining. This outcome was significantly different from all j+AV rats (n = 5), which did not exhibit any diarrheal symptoms (Table 2). Pilot studies indicated that even with repeated irinotecan doses of up to 40 mg/kg and acute doses of 80 mg/kg, j+AV rats did not experience intestinal toxicity, in contrast to j/jAV rats that were severely afflicted with diarrhea (Table 3).
Diarrhea score in j/jAV and j+AV rats (n = 5 in each group) as determined 24 h after each daily dose of irinotecan (20 mg/kg i.p.)
Scoring criteria are given under Materials and Methods.
Diarrhea scores in j/jAV and j+AV rats as determined 24 h after the last daily dose of irinotecan (20–80 mg/kg i.p.) in pilot studies
Scoring criteria are given under Materials and Methods.

Plasma profiles of irinotecan (A), SN-38 (B), and SN-38G (C) in j/jAV (♦) and j+AV (♦) rats after a 20-mg/kg dose of irinotecan i.p. (n = 5 rats per group). Each data point is the mean concentration in rats of that group with associated standard error bars.
Since toxicity differences between the groups were strikingly evident on a macroscopic basis, microscopic differences in colon and cecum integrity and composition were compared (n = 5 rats/group) (Fig. 9). In the colon and cecum of j/jAV rats, there was a significant collapse of crypts. Inflammatory cell debris and mucoid material filled the crypts. Infiltrating neutrophils were also evident. Goblet cells were widely depleted. Epithelial cells lining the crypt were fused or severely necrosed. In patchy areas, low cuboidal epithelial cells with thin cytoplasm and large nuclei lined the crypt, indicative of regeneration. In cecum and colon samples from j+AV rats, tall columnar epithelial cells lined intact villi. Goblet cells containing mucous filled the crypts. Overall, colon and cecal mucosa in j+AV rats was normal in appearance. In several j/jAV and j+AV cecum and colon samples, moderate submucosal edema was present.
Discussion
In this study, we examined the importance of intestinal UGTs in modulating irinotecan-induced diarrhea. This was accomplished by using a rat model engineered to differ from its control in intestinal UGT expression only. By using an adenoviral vector containing rat UGT1A isoforms constitutively expressed in other Wistar-derived rat strains, Gunn rats were found to have systemic SN-38 conjugation activity. After intravenous injection of adenoviruses, any virions not sequestered by Kupffer cells will be exclusively taken up by surrounding hepatocytes, precluding intestinal infection (Amalfitano, 2004). The lack of intestinal UGT expression and function in j/jAV rats was indeed shown in this study (Fig. 5). Therefore, by j/jAV rats gaining hepatic UGT activity and j+AV rats naturally possessing both hepatic and intestinal UGTs, we could test the hypothesis that intestinal UGTs are local, protective mechanisms inherent in enteric cells, the site of SN-38 cytotoxicity, and their expression is critical in the prevention of diarrhea. Because striking differences in the incidence of diarrhea and lower intestinal pathology were noted after both groups of rats were challenged with irinotecan, further experiments were conducted to ensure that the toxicity that ensued was due to a lack of intestinal SN-38 conjugation and not due to differences in systemic or intestinal irinotecan, SN-38, or SN-38G exposure.
Ideally, hepatic glucuronidation and the systemic disposition of irinotecan, SN-38, and SN-38G in the j/jAV rats would precisely mimic that in the j+AV rats. Over the entire irinotecan-dosing interval, microsomal reactions demonstrated that j/jAV rats possessed significantly higher SN-38 conjugation rates. This is most probably due to higher expression of UGT1A7 in j/jAV animals, because glucuronidation rates mimic rat 1A7 levels (Fig. 3). In addition, UGT1A7 is the major rat isoform responsible for SN-38 catalysis as determined by recombinant isoform studies (Tallman et al., 2005). Despite higher in vitro SN-38 glucuronidation rates, the exposure to SN-38G in the plasma of j/jAV rats was 2-fold lower than that in the j+AV rats. It is only speculative to compare data from these two different measures of glucuronidation. However, one explanation for this apparent discrepancy may be more efficient SN-38G basolateral transport mechanisms in the j+AV rat, which would manifest in higher plasma glucuronide levels despite lower intracellular glucuronidation rates. In rats expressing hepatic UGTs, there is a physiological need for basolateral transporters that can transport glucuronides out of the cell, such as Mrp3. Conversely, in an uninfected j/j rat, levels of these transporters may be low due to the lack of glucuronide substrates present intracellularly. Similar theories have been proposed, where Mrp3 expression is regulated by accumulating intracellular substrates (Donner and Keppler, 2001; Johnson et al., 2006). In j/jAV rats, induction of Mrp3 may lag behind UGT expression, which would explain why less SN-38G was measured in plasma. To address this, Western blots of hepatic membrane proteins from j+AV, j/jAV, and j/j rats were run. From these data, there was no observable difference in the expression of Mrp3 between these groups (data not shown). Thus, factors influencing the discordance between in vitro and in vivo measures of SN-38 glucuronidation are currently unknown. Nevertheless, the possibility that other basolateral transporters that recognize SN-38 or SN-38G might differ between groups cannot be discounted, considering that increasing UGT expression level in the j/jAV rats is not sufficient to reduce systemic levels of SN-38 (Figs. 3 and 6).

Biliary excretion (0- to 6-h collection) of irinotecan (A), SN-38 (B), and SN-38G (C) in j+AV (□) and j/jAV (▪) rats after a single 20-mg/kg i.p. dose of irinotecan. Bars represent data from a single rat and are an average of duplicate measurements.

Percentage of weight loss relative to baseline over time in j/jAV (♦) and j+AV (♦) rats (n = 5 in each group) treated with irinotecan 20 mg/kg/day i.p. × three doses. Each weight measurement was taken 24 h after the dose of irinotecan was administered. Points represent mean ± S.E. **, p < 0.01; ***, p < 0.001.
SN-38 plasma levels were found to be significantly higher in j/jAV rats. This may be a function of less SN-38 consumed by glucuronidation, as reflected in lower SN-38G levels, differences in transport of SN-38 from the hepatocyte to blood, or more efficient reabsorption of SN-38 in j/jAV animals. In a rat without functional intestinal glucuronidation, enteric cycling of SN-38 between the enterocyte and intestinal lumen would be nonexistent and more SN-38 could be reabsorbed into the blood (Jeong et al., 2005). This is not very likely, because apical secretion of SN-38 was severalfold more efficient than basolateral flux as determined in Caco-2 cells (Kehrer et al., 2000). Furthermore, the inhibition of β-glucuronidase does not reduce plasma AUCSN-38 relative to control-treated rats, indicating little enterohepatic circulation (Takasuna et al., 1998).
With the knowledge that high intestinal SN-38 concentrations may be a predisposing factor in the development of diarrhea, differences in irinotecan, SN-38, and SN-38G biliary excretion in j/jAV and j+AV rats were studied. In the intestinal lumen, irinotecan may be converted to SN-38 by carboxylesterases, and SN-38G excreted into bile is almost completely hydrolyzed to SN-38 by fecal β-glucuronidases (Takasuna et al., 1998; Khanna et al., 2000; Slatter et al., 2000). Differences in excretion of these compounds between the two groups of rats may be a confounding factor in describing the precise role that intestinal UGTs have in mediating toxicity. Although the data were derived from a small number of rats, the amounts of SN-38 and SN-38G extruded into bile were similar. The excretion of irinotecan was approximately double in the j+AV animals. From these data, j+AV rats may then have a slightly greater intestinal SN-38 burden via possible irinotecan hydrolysis. Overall, in vitro, biliary excretion and pharmacokinetic data indicate that the disposition of irinotecan and its metabolites are similar in j/jAV and j+AV rats. The small differences observed between the groups of rats with regard to these parameters probably do not explain the extraordinary degree of intestinal toxicity experienced only by the j/jAV animals.
Another factor besides differences in intestinal UGT expression that may explain the higher toxicity in the j/jAV rats is the secretion of SN-38 from the blood to the intestine. This is especially important to consider, because j/jAV rats had higher SN-38 blood levels than j/jAV rats when dosed at 20 mg of irinotecan/kg/day (Table 1; Fig. 6). However, the possibility that intestinal secretion accounts for the toxicity differences is unlikely, as determined through dose-ranging pilot studies. A 40-mg/kg i.p. dose of irinotecan gave a similar SN-38 AUC to that of a j+AV rat dosed 20 mg/kg (18.4 versus 15.2 min · μg/ml, respectively), yet the j+AV rat still did not exhibit diarrhea (Table 3). Assuming linear pharmacokinetics, an 80-mg/kg dose would produce an even higher SN-38 plasma AUC and driving force from the blood to the intestine. However, this dose did not predispose the j+AV rats to diarrhea as experienced by the j/jAV rats, indicating that a factor distinct from intestinal secretion of SN-38 is responsible for the striking toxicity differences (Table 3).

Representative micrographs (10× magnification) of j/j AV colon (A) and cecum (B) and j+AV colon (C) and cecum (D). Tissues were extracted upon euthanizing the rats, 24 h after the third dose of irinotecan, 20 mg/kg/day i.p. A description of the observed toxicity on each group of rats is given under Results.
Currently, it is unknown what level of constitutive intestinal UGT function will spare an animal from toxicity to SN-38. Heterozygote Gunn rats have only intermediate intestinal UGT1A expression relative to a Wistar rat, yet the residual activity in the former animals was sufficient to protect them from diarrhea at the doses of irinotecan used here. As with rats, patients with the lowest intestinal SN-38G formation may be those experiencing diarrhea. In humans, large differences in intestinal SN-38 conjugation are noted, and they may result from numerous factors. Various components of food, such as chrysin or octylgallate, have been found to selectively induce or potently inhibit intestinal SN-38G formation, respectively (Cummings et al., 2003; Tobin et al., 2006). Because UGT1A1 and UGT1A9 are found in the lower intestine and recognize SN-38 as a substrate, polymorphisms in these isoforms may impart low or high catalytic turnover and effect gastrointestinal adverse events (Strassburg et al., 1998; Tallman et al., 2005).
To date, low hepatic SN-38 glucuronidation has most commonly been proposed to be linked to the development of diarrhea, as evident through the establishment of the biliary index (Gupta et al., 1994). However, our data indicate that intestinal toxicity ensues regardless of hepatic UGT expression and support the following scenario. Patients with inefficient hepatic glucuronidation (i.e., Gilbert's patients) will excrete less SN-38G and similar levels of SN-38. In contrast to a patient with efficient hepatic activity, these patients will have less total intestinal SN-38 for several reasons. SN-38G is more efficiently transported into bile, and all SN-38G in the intestine is cleaved to SN-38 by β-glucuronidase (Slatter et al., 2000). Despite this, Gilbert's patients may still experience diarrhea, because of low enteric SN-38G catalysis rates. Thus, the biliary index proposed by Gupta et al. (1994) may correlate with the incidence of diarrhea in these patients, although toxicity may have little to do with systemic glucuronidation (Gupta et al., 1994). Patients with a low biliary index may also suffer adverse effects, resulting from poor intestinal turnover of a high SN-38 burden. This scenario, similarly proposed by others, may explain the poor correlation between the biliary index calculation and diarrhea (Tukey et al., 2002).
The j/jAV and j+AV rat model is being used in our laboratories to study the influence of intestinal UGTs on local toxicity. This model should be applicable to a wide range of toxic substrates that are highly glucuronidated by intestinal isoforms to inactive conjugates. In summary, the current study with SN-38 in j/jAV and j+AV rats strongly supports that intestinal UGTs function in a primary role to protect the integrity of the gastrointestinal mucosa against cytotoxic agents.
Acknowledgments
We thank Dr. Robert Kelly for SN-38G methyl ester and irinotecan.
Footnotes
-
This study was supported by National Institutes of Health Grant GM61188.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.110924.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; SN-38, 7-ethyl-10-hydroxy-camptothecin; SN-38G, SN-38 glucuronide; AUC, area under the concentration versus time curve; j/j, Gunn rat; j+, heterozygote Gunn/Wistar rat; j/jAV, Gunn rat that received adenovirus; j+AV, heterozygote rat that received adenovirus; OD, optical density; TBS-T, Tris-buffered saline with Tween; HPLC, high-performance liquid chromatography.
- Received July 16, 2006.
- Accepted September 25, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}