


Abstract
Absorption and excretion of undegradable peptides were investigated with use of octapeptides synthesized from d-amino acids.d-Tyrosine was included in each peptide to permit labeling with 125I, d-glutamic acid ord-lysine were included to vary net electric charge andd-serine or d-leucine were included to vary lipid solubility. Peptides were administered parenterally or orally to normal rats drinking 5% glucose or maltose. Forty-five percent of a lipid-insoluble, negatively charged octapeptide added to the drinking fluid in milligram quantities was absorbed from the intestine and excreted intact in urine; 90% of this peptide was recovered in urine after parenteral injection. In contrast, lipophilicd-octapeptides were largely excreted in feces, even after subcutaneous injection; the amounts excreted in feces were correlated with oil/aqueous partition coefficients. Evidence is presented that lipophilic peptides entering liver cells combine with bile salts to form hydrophilic complexes that are secreted rapidly at high concentration in bile. At physiological concentrations of bile salts (5–40 mM) and nanomolar concentrations of peptide the binding is so complete that these undegradable peptides are rapidly cleared from liver to duodenal fluid in association with the bile salts. After reaching the ileum the bile salts are reabsorbed to blood, leaving the original lipophilic peptides to be excreted in the feces from which they can be extracted, purified and identified by high-pressure liquid chromatography. These mechanisms are discussed in relation to a) the paracellular absorption of peptides and other solutes by solvent drag and b) the delivery and fate of biologically active peptides.
In a previous paper we showed that a lipid-insoluble, negatively charged octapeptide composed entirely of d-amino acids (code named DP3, see table 1) can be recovered almost quantitatively from urine and feces after either oral or parenteral administration to rats (Pappenheimer et al., 1994). More than 90% of DP3 injected intraperitoneally or subcutaneously was recovered intact in urine, and more than 45% of orally administered peptide was absorbed from the intestine and recovered intact from urine during steady voluntary ingestion with 5% glucose. Recovery of this octapeptide was independent of ingested loads up to at least 50 mg (65 μmol), and it seemed probable that the mechanism of absorption was solvent drag in fluid absorbed through tight junctions and intercellular channels that were dilated by the glucose (Pappenheimer and Reiss, 1987; Madara and Pappenheimer, 1987;Atisook and Madara, 1991; Pappenheimer and Madara, 1993). In contrast, it will be shown in this paper that a lipid-soluble, positively charged octapeptide (code named DP1, table 1) is largely excreted in the feces, even after subcutaneous injection. It therefore appears that efficient paracellular intestinal absorption of these undegradable peptides and their excretion in feces or urine depends on their lipid solubility or their net charge or both.
Oil/aqueous partition coefficients of 125I-labeledd-octapeptides
Undegradable peptides composed of d-amino acids are well suited to investigation of these problems because their composition can be adjusted to alter charge and lipid solubility independently without significant alteration of molecular size. The problem is of special importance to theories of paracellular intestinal absorption of nutrients (Pappenheimer and Reiss, 1987; Pappenheimer, 1993;Pappenheimer and Madara, 1993; Karasov and Cork, 1994; Gardner, 1995), but it is also of practical importance for peptide delivery systems, especially because several peptides composed partially or entirely ofd-amino acids have recently been shown to be potent, long-acting substitutes for naturally occurring peptide hormones or antibiotics (Bauer et al., 1982; Lundin and Vilhardt, 1986;Fuessl et al., 1987; Dooley et al., 1994;Merrifield et al., 1995; Quillan et al., 1995). In the present paper we show that lipid-soluble d-peptides having ionized amino groups and capable of entering liver cells are secreted rapidly from blood to duodenal fluid in association with bile salts. Ingested, positively charged peptides can also combine with bile salts and/or albumin in duodenal fluid and are therefore prevented, at least partially, from entering tight junctions. Of the fived-octapeptides tested, only the most hydrophilic (DP2 and DP3) were absorbed efficiently by paracellular solvent drag between absorptive cells of the jejunum.
Methods
Synthesis and chromatographic characterization ofdoctapeptides.
The following peptides with molecular weights in the range 783 to 877 daltons were synthesized from d-amino acids as described previously (Pappenheimeret al., 1994) to provide a spectrum of lipid solubilities and net electric charges at pH 7.4: lys-ala-leu-ala-leu-tyr-leu-ala (DP1), lys-ala-ser-ala-ser-tyr-ser-ala (DP2), glu-ala-ser-ala-ser-tyr-ser-ala (DP3), lys-ala-lys-ala-leu-tyr-leu-ala (DP4) and glu-ala-leu-ala-leu-tyr-leu-ala (DP5). Composition of the unlabeled peptides was confirmed by amino acid analyses and/or by mass spectrometry. After labeling with 125I, the peptides were applied to octyldecyl silica gel cartridges at pH 2 or pH 4 (C-18 Sepak, Waters Co., Milford, MA) and then eluted with graded alcohol-0.01 M TFA mixtures in steps of 10% or 20% alcohol, usually with 1 ml of each alcohol mixture. Each peptide had a characteristic elution pattern. Approximately 95% of the 125I in each sample of labeled peptide was retained by the Sepak gel from which it could be subsequently eluted. Labeled peptides were also subjected to gel filtration on a G15 Sephadex sizing column (inside diameter 1.6 cm, length 60 cm) with 0.01 M PO4 buffer, pH 6.8, as elution vehicle at a flow rate of 0.6 ml/min. The peak elution volumes after Blue Dextran or bovine serum albumin were 19, 20, 32 and 62 ml, respectively, for peptides DP1, DP2, DP3 and free NaI. DP4, with two lysine moieties, failed to elute at pH 6.8 but eluted with a peak at 15 ml with 0.01 M borax-PO4 buffer at pH 8.6. The G15 column was therefore useful for separating these labeled peptides from protein and from free iodide. Most of the 125I, which failed to be retained by the Sepak gel (1–6% of the total 125I), was in the form of free iodide as revealed by the G15 Sephadex filtrations.
The labeled peptide eluates from Sepak were subjected to final purification and detailed characterization on HPLC as described previously (Pappenheimer et al., 1994).
Distribution of peptides between oils and aqueous buffers.
Classical studies by Collander and Barlund in 1933 correlated the permeability of biological membranes to organic solutes with their solubilities in olive oil. However, oil/aqueous partition coefficients have not, to our knowledge, been used previously for physiological or chemical studies of peptides. In the present investigation we used partition coefficients not only for correlation with permeability but also as a means of measuring the binding of peptides with bile salts and with serum albumin. In preliminary experiments we found that peptides containing the lipophilic amino acid leucine were very soluble in vegetable oils consisting of complex mixtures of fatty acid esters (commercial olive oil or safflower oil); they were also soluble in “mineral” oil consisting of hydrocarbons (clinical grade). The following procedure was developed for measurements of partition coefficients. Labeled peptides in pH 7.4 buffer were brought to ionic strength 0.15 M with NaCl. Aliquots (1.5–2 ml containing at least 5000 cpm of labeled peptide) were pipetted into 12 × 100 mm disposable test tubes. An equal volume2 of oil (safflower, olive or mineral) was layered over the aqueous phase and the tubes equilibrated to 38°C in a water bath. At zero time, the tubes were removed from the bath, mixed for 30 s (Vortex) and returned to the bath for 30 s. This mixing procedure was repeated 10 times and produced smooth oil/aqueous emulsions. After 10 min the emulsions from each tube were poured into two 1.7-ml centrifuge tubes and centrifuged for 5 min at 10,000 rpm (Eppendorf). One-milliliter samples of the separated phases were drawn into glass tuberculin syringes and discharged into tubes for γ-scintillation counting. For peptides that are poorly soluble in oil, care must be taken to obtain clear samples of the oil phase without even slight contamination with the aqueous phase. The separation of the two phases is usually complete but there is often a cobweb precipitate at the interface between phases. Some of the peptide adheres to the cobweb precipitate so that the total radioactivity recovered from both phases may be less than 100%, especially for those peptides that are very soluble in the oil phase. For purposes of this investigation we define the partition coefficient (α) as the ratio of counts per minute per milliliter in the oil phase to that in an equal volume of the aqueous buffer phase. However, in aqueous solutions containing hydrophilic solutes that bind with the peptide the ratio of counts may be diminished in proportion to the binding, and under these circumstances the oil/aqueous ratio of counts is defined as θ to distinguish it from the true α of the free peptide. θ/α will be used in the present paper to calculate binding constants as described below in the section on binding of peptides to bile salts.
Animals, experimental protocols and operative procedures.
Adult male or female white rats weighing 200 to 350 g were maintained on about 15 g/day of chow pellets with water ad libitum. This limited food intake was sufficient to maintain body weight but made the rats eager to ingest 5% glucose or maltose when they were placed in metabolism cages for collection of urine and feces. The 5% carbohydrate (CHO) was supplied for two reasons: 1) to stimulate a copious absorption of fluid from the jejunum with associated dilation of intercellular junctions and absorption of peptides and other solutes by solvent drag (Pappenheimer, 1990;Pappenheimer et al., 1994) and 2) to stimulate a copious flow of urine for efficient collection of the labeled peptides. Two to ten percent glucose, 4 to 6% maltose or 4 to 5% egg albumen were all effective in producing large voluntary fluid intakes with associated steady state absorption and excretion rates of fluid containing peptides, but after preliminary trials we standardized on 5% glucose or maltose. In the absence of such nutrients the fluid intakes were too small to accommodate the quantities of hydrophobic peptides and creatinine required for these measurements. Fluid ingested from weighed bottles contained 0.25 to 1% creatinine and about 104cpm/ml 125I peptide made up in 5% CHO. In some experiments, 10 to 50 mg of unlabeled peptide were added to the ingestate, but the percentage recoveries from urine or feces were independent of load within this range as described previously (Pappenheimer et al., 1994). Creatinine was included in the ingestate because its subsequent excretion in urine provided an independent measure of paracellular absorption from the intestine (Dominguez and Pomerene, 1945a,b; Pappenheimer, 1990; Pappenheimeret al., 1994). Experiments were started in late afternoon and continued overnight; thereafter, the drinking fluid contained only 5% CHO and the experiment was continued for 3 to 6 days until 80 ± 10% of the 125I had been recovered in urine plus feces. A small amount of solid food (4–8 g pellets) was added to the diet after 24 hr to promote formation of solid feces. Recoveries of peptides after subcutaneous injections were not significantly different from those after intraperitoneal injections; therefore, both are included together under the heading “parenteral.”
Intravenous infusions and segmental intestinal perfusions were carried out under deep surgical anesthesia with Na-pentothal. Duodenal or ileal segments (3–6 cm long) were perfused from a syringe pump while labeled peptide in Ringer-HCO3 was infused into an external jugular vein at 1.4 ml/hr. Control perfusates were collected without infusion of peptides. The perfusates were collected through PE 205 tubing inserted 6 to 8 cm distal to the pyloric valve; an inner PE 60 tube connecting with the perfusion syringe was placed with its tip 3 to 5 cm beyond the orifice of the collecting tube. At an inflow of 0.6 ml/min the bile was diluted about 30-fold in the outflow (see “Results”). Samples of heart blood and tissues were removed when the animals were sacrificed at the end of each perfusion.
Extraction, purification and identification of labeled peptides from urine, intestinal perfusates or feces.
Urine samples or intestinal perfusates were ordinarily titrated to pH 2 and passed through Sepak cartridges from a syringe pump at 0.6 ml/min. Urine samples containing high concentrations of creatinine also contained high concentrations of carbonates associated with the creatinine; titration to pH 2 therefore released large volumes of CO2, and care was needed to remove foam before passing the samples through the Sepak cartridges. In experiments on binding of peptides with glycocholic or taurocholic acids the Sepak filtrations were carried out at pH 4 to prevent precipitation of the bile salts3 at low pH. Peptides retained in the cartridge were eluted with alcohols as described under “Synthesis and Chromatographic Characterization of dOctapeptides,” and the elution pattern was compared with that of the original peptide (table 4). The matching radioactive eluate was subjected to G15 filtration to remove high molecular weight contaminants, the fractions containing the peptide were combined and again subjected to Sepak concentration. In some experiments the intestinal perfusates were subjected to ultrafiltration to remove solutes of molecular weight greater than about 10,000 daltons (Amicon UM10 filters, Millipore Co., Lexington, MA). Eluates from the second Sepak were subjected to HPLC for final identification.
Elution4-a profiles from Sepak cartridges, pH 4.0
Peptides were extracted from feces with GITC by the technique developed by Too and Maggio (1995) for extraction of peptides from tissues. The GITC solution consisted of 5.9 g GITC, 0.3 g NaOAc in 9 ml distilled water. Feces (300 mg) were macerated with 5 ml of GITC solution. Lipids were extracted with equal volumes of CHCl3/isopropyl alcohol (2:1 v/v). Approximately 15% of the most lipid-soluble peptide (DP1) was lost to the CHCl3phase, and 50% of the original radioactivity in the feces was recovered in the GITC extract. Most of the remaining activity was in particulate matter. The aqueous GITC extract was diluted 5-fold and titrated to pH 2.0 for Sepak filtration. The eluates from Sepak were subjected to G15 filtration and a second concentration on Sepak before final identification with HPLC.
Results
Oil/aqueous partition coefficients, α.
Table1 summarizes the results obtained with safflower, olive and mineral oil at pH 7.4. The peptides are listed from left to right in descending order of their oil/water partition coefficients. The lower case abbreviations for the amino acids signify that they were the dextro isomers. Peptides containing leucine were highly soluble in safflower oil, the most soluble being DP1 (α = 1.05). When expressed relative to DP1 all peptides had approximately the same relative solubilities in all three oils; average values relative to DP1 are shown in the last row of table 1. Because mineral oil is an inert mixture of hydrocarbons this result indicates that partition coefficients of these peptides in vegetable oils do not depend on special affinity with fatty acids or their esters and any one of these oils could be used equally well for correlations with membrane permeability. In the present paper we use the mean values of α relative to DP1 found in all three oils and summarized in the last row of table 1.
Excretion of peptides after single parenteral injections or after steady ingestion with 5% CHO.
Table 2 shows recoveries of 125I after subcutaneous or intraperitoneal injections of labeled peptides (columns 4–5) or after ingestion overnight with 5% glucose or maltose (columns 7–8). The peptides are listed in descending order of their solubilities in oil (column 3). Volumes of fluid ingested each night were in the range 50 to 100% of body weight, and the volume of urine produced was about 75% of the volume ingested. Forty to 60% of ingested creatinine was recovered in urine. The volumes ingested and excreted, together with values for absorption-excretion of creatinine, are not included in table 2 because they do not differ significantly from those given in detail previously, and we need only emphasize here that the rats drank and urinated so frequently throughout the night that the rates of ingestion and urinary excretion of both creatinine and the peptides reached steady states as illustrated in figure 1 of our previous publication (Pappenheimeret al., 1994). The values for urinary recovery of peptides have been corrected for nonpeptide 125I in both the administered loads and the samples of urine, i.e., they refer to the 125I retained by Sepak cartridges and subsequently eluted in the appropriate alcohol fraction as described under “Methods.” For this reason the uncorrected (total) recoveries of 125I listed in columns 6 and 9 are slightly greater than the sum of the corrected urinary and fecal values. Most of the labeled peptides excreted in urine were recovered within 12 hr of a single parenteral injection or within 12 hr after overnight ingestion of labeled peptides in the drinking fluid. In contrast, 3 to 6 days were required for excretion of the fraction found in feces. The small amounts of 125I that were excreted in urine after 24 hr were mostly in the form of iodide or other low molecular weight compounds as revealed by passage through Sepak cartridges or by filtration through the G15 Sephadex sizing column.
Recovery of peptides and of 125I after administration to normal rats

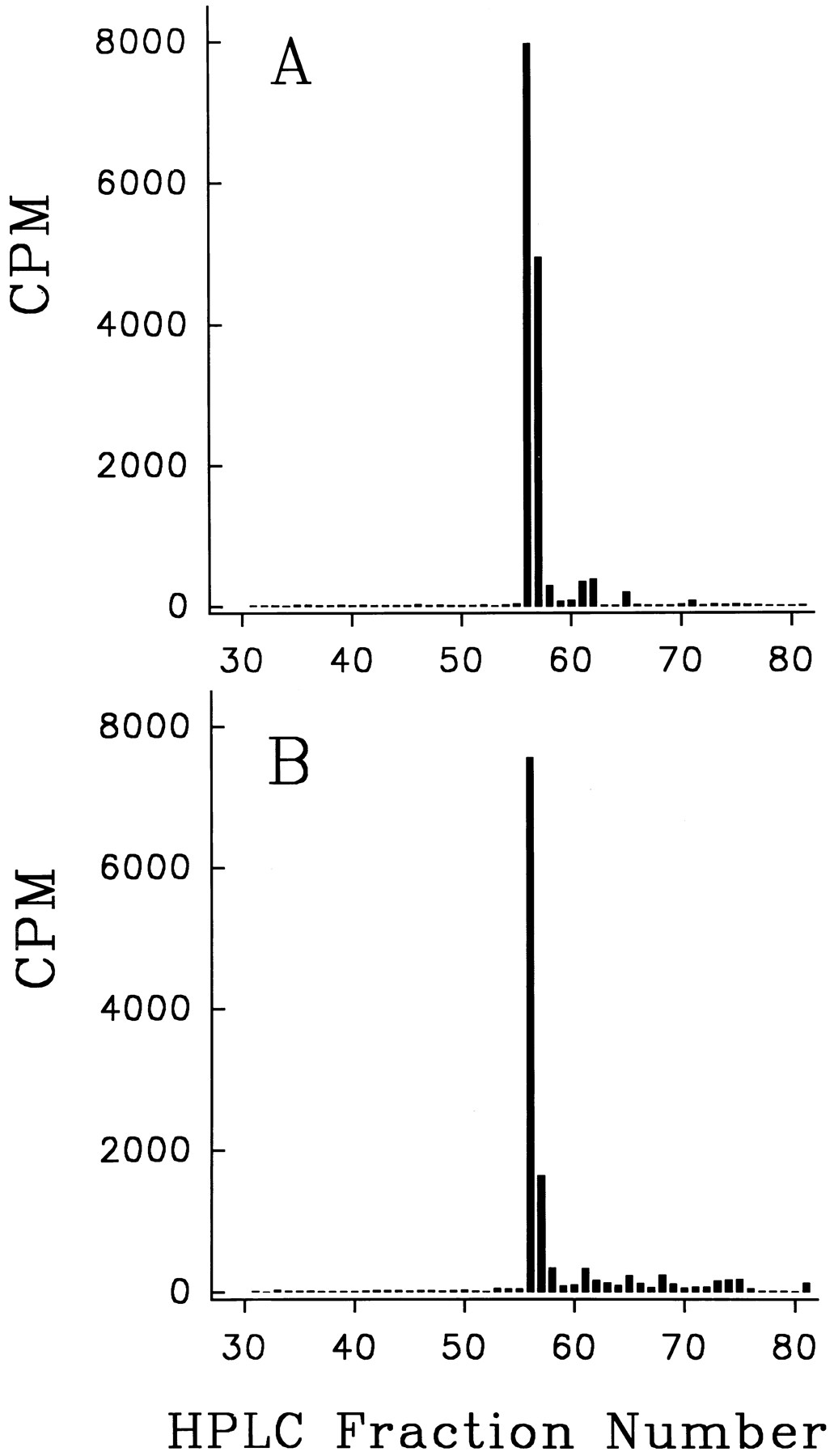
Reverse-phase HPLC of original labeled DP1 (A) and of the label isolated from feces (B). A 345-g rat was injected subcutaneously with 430 μmol of labeled DP1. During the next 3 days 8% of the label was excreted in urine and 41% in feces. The feces were extracted with GITC and the labeled component concentrated and purified by the Sepak and G15 Sephadex procedures described under “Methods.” An aliquot of the final Sepak eluate was used for the HPLC shown in panel B.
Although retention in Sepak cartridges followed by elution with alcohol mixtures provides an approximate measure of the fraction of original labeled peptide in any given sample, it does not provide rigorous evidence that all the 125I in the eluates is in the form of the original labeled peptide. For this reason the eluates from the first Sepak were subjected to G15 Sephadex filtration to remove high molecular weight impurities, and the active fractions from G15 were again taken up and eluted in concentrated form from a second Sepak cartridge. Samples from this second Sepak were then used for final identification with HPLC. Results for the most hydrophilic peptide DP3 have already been published and they showed that the label was in the form of the original labeled peptide (Pappenheimer et al., 1994). Similar results have now been obtained from the most lipophilic peptide of the series (DP1), which is mainly excreted in feces after either oral or parenteral administration (table 2, row 1). The HPLC elution profile of the labeled material extracted and purified from feces did not differ significantly from that of the original peptide as illustrated in figure 1, A and B.
Urinary and fecal recoveries of peptides are plotted in figure2, A and B, as a function of their relative solubilities in oils. DP4 is omitted from figure 2 because its additional ionized amino groups (table 1) promote binding with bile acids in duodenal fluid, thus introducing a disproportionate restriction to absorption through tight junctions (see “Binding of DP1 and DP5 with Bile Salts”). The fact that 74% of parenterally injected DP4 was recovered from urine (table 2) also suggests that the three ionized amino groups restricted its penetration into liver cells despite its lipid solubility.

Effects of lipid solubility on absorption and urinary excretion of d-octapeptides by normal rats drinking 5% glucose or maltose. The standard errors of each point were less than ±5% as shown in table 2. Data from peptide DP4 are not included because its three ionized amino groups cause disproportionate restriction of paracellular absorption and entrance into liver cells (see text).
The results summarized in table 3 and figure 2 show that the most hydrophilic peptides (DP2 and DP3) were excreted almost quantitatively by the kidneys, and little or none was recovered from feces even after 6 days. Of these peptides 31 to 48% were also recovered from urine after ingestion with 5% CHO, thus proving that substantial quantities of these inert, undegradable octapeptides can be absorbed from the intestinal tract. The mechanism of absorption is presumably via solvent drag in fluid absorbed through intercellular junctions of the jejunum as described in detail previously for DP3 (Pappenheimer et al., 1994) and for other solutes (Pappenheimer and Reiss, 1987; Pappenheimer, 1990; Perezet al., 1993; Karasov and Cork, 1994).
Oil/aqueous partition coefficients of DP1 and its complexes in intestinal perfusates and in bile salt solutions in vitro 3-a
In contrast, the most lipophilic peptides (DP1 and DP5) were largely excreted in feces, even after parenteral injections. The only known pathway for rapid transport of solutes from blood to the lumen of the gastrointestinal tract is via the enterohepatic circulation and this would require conversion of these highly lipid-soluble peptides to more polar forms to be secreted in high concentration to bile. To examine this hypothesis experimentally we measured the oil/aqueous partition coefficients of labeled compounds secreted into the duodenum during intravenous infusion of the labeled peptides in anesthetized rats.
Secretion of a lipid soluble d-octapeptide (DP1) from plasma to bile and intestinal fluids.
In preliminary experiments, five mice and two rats were injected parenterally with labeled DP1 (10–20 nmol/100 g b.wt.); after 1 to 2 hr the animals were anesthetized for removal of tissues and tissue fluids. More than 50% of the injected counts were recovered from washings of the intestinal tract. The concentration of label in the washings was 10 to 25 times that of plasma in heart blood drawn at the time of sacrifice. The concentrations of label in samples of gallbladder bile from mice were more than 100 times the concentrations of plasma. Although these preliminary experiments left little doubt that the label on parenterally administered DP1 could be rapidly cleared from plasma and concentrated in bile and intestinal fluid, it remained to be shown how the label could be equated with the original peptide. Moreover the label was cleared from plasma so rapidly that the relation of its concentration in a single plasma sample to that in bile was only of qualitative significance. To investigate the phenomenon more quantitatively and if possible to elucidate the mechanism of transport from plasma to bile we carried out steady-state intravenous infusions of DP1 combined with perfusion of the duodenum in anesthetized rats.
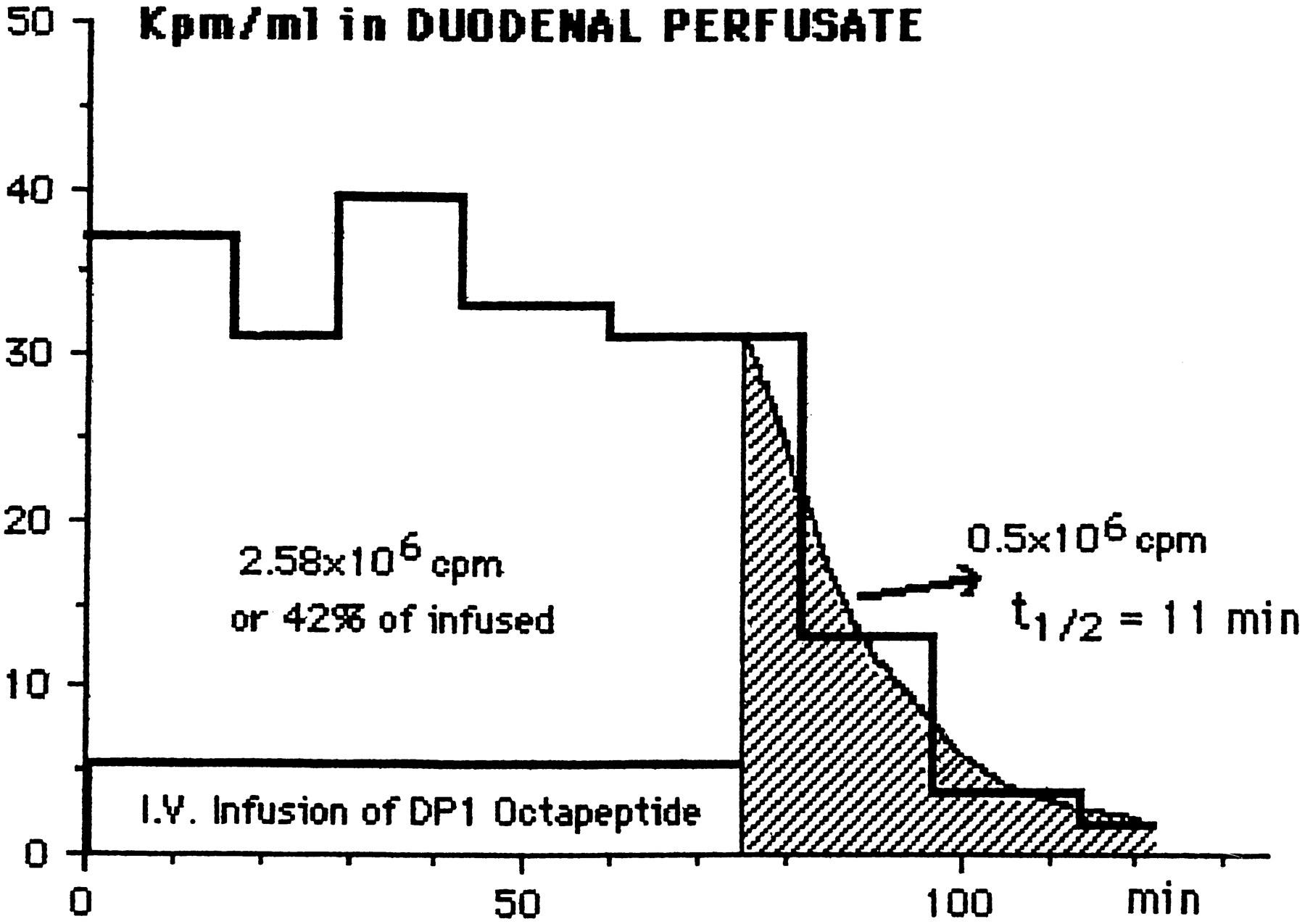
Figure 3 illustrates one experiment. After an i.v. priming dose of 0.66 × 106 cpm, the labeled DP1 was infused at the rate of 82 × 103 cpm (10 nmol of peptide)/min for 75 min (total = 6.15 × 106 cpm, 0.75 μmol). At the same time a 3-cm segment of duodenum opposite the bile duct was perfused with Ringer-bicarbonate at the rate of 0.6 ml/min. The average rate of secretion of the label to the duodenal perfusion fluid was 42% of the infusion rate, and the average concentration of label in the perfusate was 53 × 103cpm ml−1. When the infusion was discontinued after 75 min the plasma and tissue fluids were cleared of the label with a half-time of 11 min, and another 0.5 × 106 cpm were released into the perfusate within 25 min. If it is granted that this 0.5 × 106 cpm is derived from plasma and extracellular fluids (about 40 ml in the 240-g rat), then the concentration of label in plasma at the time the infusion was stopped was 12.5 × 106 cpm/ml. Thus the concentration in the duodenal perfusate (53 × 103 cpm ml−1) was more than four times the concentration in the plasma. The volume flow of bile in anesthetized rats or from perfused rat livers is about 20 μl/min per 300-g rat (Archdeacon et al., 1954;Despopolous, 1966; Berthelot et al., 1970; Boyer, 1971;Whelan and Combes, 1971; Harbison and Wood, 1978) or 30-fold less than the perfusion rate (600 μl/min). Therefore, the concentration of label in original bile during the steady-state infusion-perfusion was 120-fold that in plasma, thus confirming the high concentration ratio estimated from the preliminary (non-steady state) experiments on mice and rats described at the beginning of this section. In four similar experiments on rats the secretion rate of labeled DP1 from blood to duodenal perfusion fluid averaged 38 ± 10% of the infusion rate. In a single experiment on an anesthetized rabbit the bile was collected directly from the duct at the rate of 0.1 ml min−1 for 1 hr while labeled DP1 was infused intravenously. The average concentration of label in bile was 64 times that of its average concentration in arterial plasma, thus confirming in the rabbit the high concentration ratios found in mice and rats.

Secretion of DP1 octapeptide from blood to duodenum. Labeled DP1 was infused i.v. at the rate of 82 cpm/min (10 nmol/min) in a 240-g anesthetized rat whereas the duodenum was perfused with Ringer-bicarbonate at the rate of 0.6 ml/min. The 125I label was recovered in the perfusate at an average rate of 42% of the infusion rate for 75 min. After stopping the infusion, the plasma and extracellular fluid were cleared of the label with a half-time of 11 min. Calculations described in the text indicate that the peptide was secreted to bile in combination with bile salts at a concentration about 120 times the concentration of plasma.
Oil/aqueous partition coefficients in intestinal fluids.
Table3 shows that the oil/aqueous partition coefficients of the compound secreted into duodenal perfusates (row 2) were less than 7% of those of the original DP1 peptide (row 1). The mean θ/α for all three oils was 0.04, which indicated that 96% of the label was in the aqueous phase. This result supports the hypothesis that the lipid-soluble DP1 is converted to a hydrophilic form in the liver and secreted at high concentration to bile. The concentration of label in ultrafiltrates of duodenal perfusates (Amicon UM10) is more than 80% of that in the perfusate, and θ in the ultrafiltrates is less than 10% of α as shown in row 3 compared with row 1. Therefore, most of the hydrophilic labeled compound in duodenal perfusates has a molecular weight of less than 10 kdaltons. It is also possible, however, that duodenal fluid normally contains substances that can react with DP1 to form a hydrophilic compound or compounds. Row 4 of table 3 shows that this is in fact the case: addition of labeled DP1 to control duodenal perfusates in vitro reduces θ by amounts similar to those found in samples of duodenal fluid collected during i.v. infusions of DP1 in vivo (row 2). Ultrafiltrates of these perfusates also have a low θ as shown in row 5.
The results summarized in rows 1 to 5 of table 3 could be explained if the lipid-soluble labeled DP1 entered liver cells from blood and there combined with bile salts to form a hydrophilic complex that is secreted at high concentration to bile on the same Na-dependent carrier as the bile salts. This hypothesis is supported by results shown in rows 6 and 7 of table 3. Labeled DP1 added to pure solutions of taurocholic or glycocholic acids (2.5 mM, pH 7.4) in vitro becomes hydrophilic and by amounts comparable with the values found in duodenal perfusates (rows 2 and 4). These results also suggested that θ might be used to investigate the binding of peptides to bile acids.
Binding of DP1 and DP5 with bile salts.
Figure4 shows the binding of DP1 as a function of concentration of taurocholic or glycocholic acids in buffer at pH 7.4, ionic strength 0.15 M. The ordinate is θ/α, where α is (cpm)oil/(cpm)aqueous of the free peptide in the absence of bile salts and θ is (cpm)oil/(cpm)aqueous at the concentrations of bile salts shown on the abscissa. The smooth curve drawn through the experimental points is a theoretical curve relating θ/α to the mass action affinity constant, Ka
. The theoretical curve is derived as follows: I. Let α = [P]o/[P]aq where [P] = concentration of free peptide and subscripts o and aq refer to oil and aqueous phases, respectively. Let a = [PB]o/[PB]aq, where [PB] = concentration of peptide/bile salt complex. Let θ = (CPM)oil/(CPM)aqueous = {[P]o + [PB]o}/{[P]aq + [PB][ind]aq} whence:

Binding of DP1 (kalalyla) to bile salts. Oil/aqueous partition coefficients, θ/α, as a function of bile salt concentrations at pH 7.4. ○, taurocholate and safflower oil; +, taurocholate and mineral oil; •, glycocholate and mineral oil. The solid line is the theoretical binding curve (equation 3 in the text) for an affinity constant of Ka = 0.14 mM.
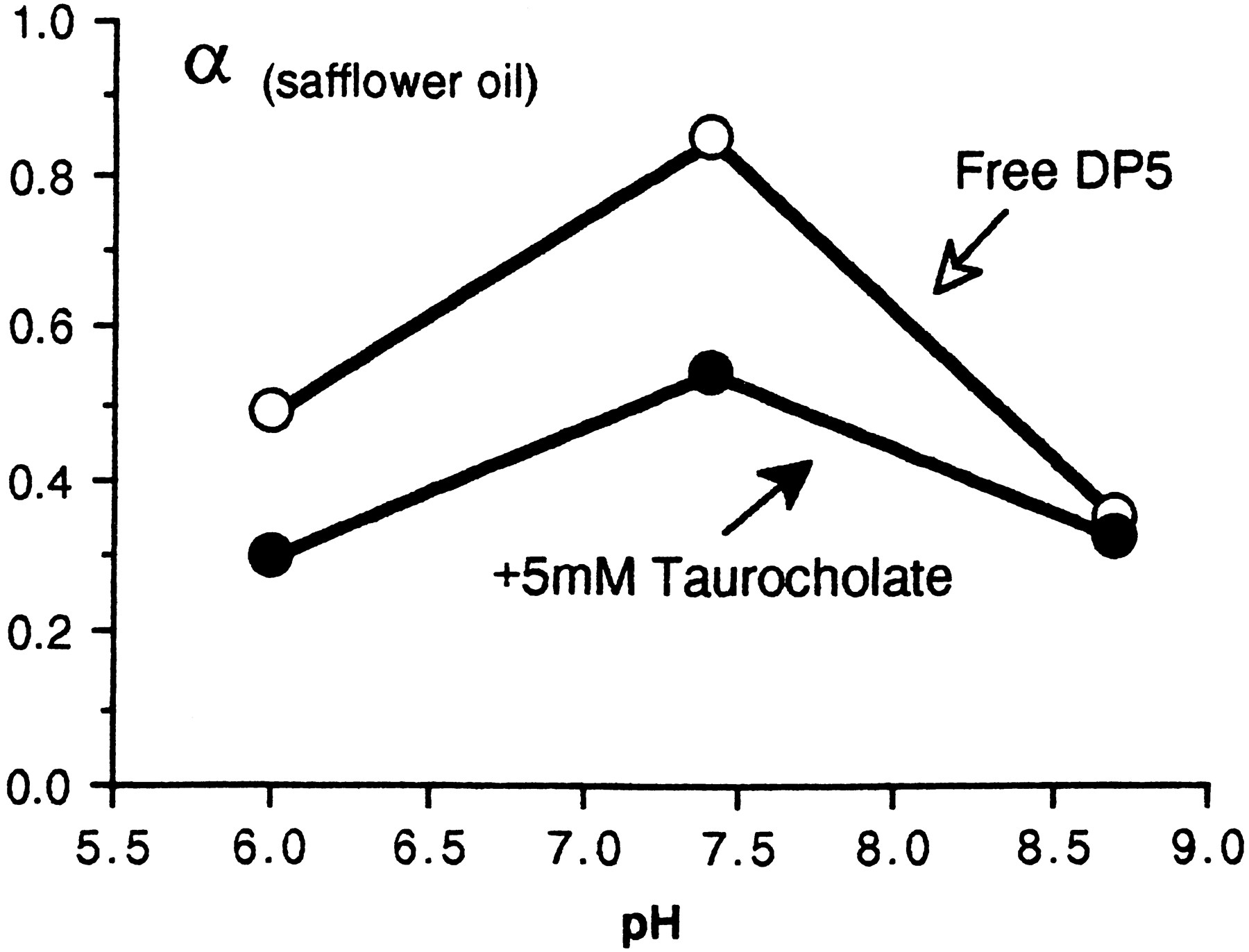
Presumably the high affinity of DP1 for bile salts derives in part from the ionized amino groups on the N-terminal lysine which bind electrostatically with the acidic groups on taurocholic or glycocholic acids. In the rat the primary bile salt is taurocholate (Berthelotet al., 1970) which may be present in concentrations exceeding 40 mM (Strange, 1984). Given aKa of 0.14 mM as in figure 4 it follows that more than 98% of DP1 will be bound to taurocholate at a normal pH of 7.4. In DP5 (which is also lipid soluble but has a net negative charge at pH 7.4) there are fewer ionized amino groups, the binding with taurocholic acid is less complete and its secretion into bile after parenteral injection is less efficient than that of DP1 as shown in table 3. At pH 8.5 in vitro the ionization of amino groups on DP5 is suppressed and there is no detectable binding with taurocholate as measured by θ/α (fig. 5).

Effect of pH on safflower oil/aqueous partition coefficient of DP5 (ealalyla). At pH 6.0 and 7.4 the terminal amino group on glutamic acid is partly ionized and available for binding with taurocholate to form a more hydrophilic complex. At pH 8.7 the amino group is unionized and there is no significant change of oil/aqueous partition coefficient in the presence of taurocholate.
Elution profiles from Sepak cartridges.
At pH 4 more than 95% of the label is retained when solutions of labeled DP1 in buffer, in duodenal perfusates or in solutions of bile salts are applied to Sepak C18 cartridges. Elution profiles using graded alcohols are summarized in table 4. The elution profiles of label retained from duodenal perfusates or from DP1-taurocholate solutions are shifted to the right of the original and they are virtually identical as would be expected if DP1 were combined with taurocholic acid in rat bile. The profile from DP1-glycocholate is shifted slightly to the left of the original, and glycocholate is therefore not the principal carrier of DP1.
DP1 in ileal perfusates.
Segments of lower ileum, 3 to 10 cm in length, were perfused as described in “Secretion of a Lipid-Soluble d-Octapeptide (DP1) from Plasma to Bile and Intestinal Fluids.” Labeled DP1 was added to samples of the perfusates and its oil/aqueous partition coefficient was measured; results are summarized in table 3, row 8. The average value of θ/α was 0.3 as compared with 0.04 in duodenal perfusates. In ultrafiltrates of ileal perfusates θ/α averaged 1.2 ± 0.1 (row 9) as compared with 0.09 in ultrafiltrates from duodenal perfusates (row 3). This striking difference is consistent with the hypothesis that the peptide-bile salt complex dissociates in the ileum as the bile salts combine with their high affinity reabsorptive carrier, leaving the original peptide to be excreted in the feces. This would explain the recovery of intact DP1 from extracts of feces.The partial reduction of θ/α in ileal perfusates (row 8) may be ascribed to proteins in ileal fluid because DP1 binds to serum albumin as shown in row 10.
Discussion
Polypeptides in intestinal chyme are normally hydrolyzed to amino acids by extracellular peptidases anchored to the microvilli of the absorptive cells (Matthews, 1975; Gardner, 1984, 1995; Semenza, 1986;Lee, 1989; Ugolev, 1989; Taki et al., 1995); the subsequent transepithelial absorption of amino acids is therefore closely coupled to enzymes in the immediate vicinity of cell junctions and membrane carrier proteins. Although small quantities of certain di- or tripeptides can be transported across epithelial membranes by ion-coupled carriers, even these peptides may be subject to hydrolysis within absorptive cells or in blood (Ganapathy and Leibach, 1985;Gardner, 1995; Meredith and Boyd, 1995). Only a fraction of 1% of natural oligopeptides that enter the intestinal mucosa ordinarily reach the systemic circulation, and hydrolysis by membrane-bound peptidases is generally believed to be one of the protective defences against ingested antigenic peptides. In addition, it is widely believed that tight junctions between absorptive cells effectively block passage of solutes in the molecular weight range of oligopeptides. Although these mechanisms are of importance for protection against antigenic peptides, they are also inconvenient barriers to the oral delivery of clinically useful peptide drugs and hormones.
Recent investigations have shown that during rapid fluid absorption associated with Na+-coupled transport of hexoses in the small intestine, the tight junctions become dilated and allow the passage of solutes between absorptive cells (Madara and Pappenheimer, 1987; Pappenheimer, 1990, 1993; Madara, 1991; Pappenheimer and Volpp, 1992; Perez et al., 1993; Karasov and Cork, 1994;Pappenheimer et al., 1994; Yen and Lee, 1995; Fricker and Drewe, 1995). In normal mammals and birds drinking large quantities of soluble carbohydrates most of the glucose formed by membrane digestion is absorbed by this mechanism. The equivalent channel width of tight junctions during absorption of glucose by the small intestine of the rat has been estimated to be 50 Å (Pappenheimer and Reiss, 1987), and theoretically there should be little restriction in such channels to the passage of solutes with molecular weights up to 1000 or more, including any polypeptides that escape hydrolysis. In our preliminary investigation (Pappenheimer et al., 1994) we found that more than 60% of creatinine and 45% of an undegradable octapeptide of molecular weight 784 (DP3) were excreted intact in the urine of normal rats during steady ingestion with 5% glucose. For the present, more extensive investigation we synthesized five such octapeptides having approximately the same molecular size, but differing greatly in their lipid solubilities and electric charge. Substantial quantities of all these peptides (5–45% of ingested load) were excreted intact in urine during steady voluntary ingestion overnight with 5% glucose or maltose. Because it is improbable that intestinal absorptive cells have developed mechanisms for transcellular transport of milligram quantities of these synthetic peptides, these results leave little doubt that they were absorbed paracellularly, presumably by solvent drag in the rapid fluid absorption induced by ingestion of large volumes of soluble carbohydrates. There was no reason to suppose, however, that the lipid-soluble octapeptides would be restricted from entering the same paracellular channels as the hydrophilic peptides anda priori we expected that the lipid-soluble peptides might be absorbed transcellularly as well as paracellularly and thus be more efficiently absorbed from the intestine. It came as a surprise, therefore, to find that the most lipid-soluble peptides were the most poorly absorbed and that instead their excretion in feces was approximately proportional to their lipid solubilities (fig. 2).
Reasons for the poor (net) absorption of lipid-soluble peptides became evident when it was discovered that they were rapidly excreted to bile after intraperitoneal or subcutaneous injection. The mechanism of their transport to bile, namely bound in dissociable form to bile salts, appears to be new to the field of peptide transport and indeed to epithelial transport in general. The bile salts are known to be concentrated and secreted rapidly on Na-dependent protein carriers in liver cells (Hagenbuch et al., 1991). It appears from the present investigation that the bile salts, in turn, can act as carriers for those peptides that can enter liver cells by virtue of their lipid solubility. Essentially, these peptides ride “piggyback” on the bile salts (mainly taurocholate in the rat). The efficiency of this transport mechanism in rats is such that during steady intravenous infusions of DP1 40% or more of this lipid-soluble peptide is secreted to bile. The affinity of taurocholate for DP1 at pH 7.4 is greater than for DP5, presumably because DP5 has fewer ionizable amino groups. For both peptides the affinity, measured in vitro by the oil/aqueous distribution technique, is decreased as the number of ionized amino groups is decreased at high pH as illustrated for DP5 in figure 5. Peptides can also be linked to bile salts covalently by chemical synthesis in vitro, and such conjugates do not interfere with carrier-mediated transport of bile salts in the ileum (Kramer et al., 1994). However, these conjugates are not synthesized in vivo, nor are they dissociable.
The relevance of this investigation to the design of peptides for oral delivery assumes greater importance because of recent discoveries showing that certain peptides composed wholly or in part ofd-amino acids are potent agonists of receptors for naturally occurring peptide hormones of clinical interest. These include a d-hexapeptide agonist of opioid receptors (Dooleyet al., 1994), the d-isomers of the antibacterial peptides cecropin and mellitin (Merrifield et al., 1995), a biologically active octapeptide region of Somatostatin (SM20-5-995) modified to include two d-amino acids (Bauer et al., 1982; Fuessl et al., 1987;Fricker and Drewe, 1995), the d-peptide antagonist to α-melanocyte-stimulating-hormone (Quillan et al., 1995) and d-arginine vasopressin (Lundin and Vilhardt, 1986). It seems likely that other biologically active peptides will be modified for clinical use by substitution or addition of d-amino acids, and the basic factors determining their absorption and excretion described in the present paper should be applicable to the design and use of such peptides.
Acknowledgments
The late Professor C.Richard Taylor provided laboratory space, facilities and encouragement in support of this research at the Concord Field Station. We are indebted to Dr. Charles Dahl in the Department of Biological Chemistry and Molecular Pharmacology at Harvard Medical School for synthesizing the d-octapeptides and for confirming their composition. We are grateful to Professor J.T. Edsall for reviewing the manuscript.
Footnotes
-
Send reprint requests to: J. R. Pappenheimer, Concord Field Station, Harvard University, Old Causeway Road, Bedford, MA 01730.
-
↵1 This work was supported in part by a grant to J.R.P. from the American Heart Association. Labeling and HPLC of peptides were supported by National Institutes of Health Grant GM 15904 to J.E.M.
-
↵2 Theoretically, the equilibrium partition coefficient (α), should be independent of relative volumes of the two phases. In practice we find that α increases almost in inverse proportion to the ratio of oil/aqueous volumes. We have no explanation for this phenomenon, which occurs in both vegetable and “mineral” hydrocarbon oils. For purposes of the present paper this unexpected property is inconsequential so long as the same volume ratio is used throughout.
-
↵3 We use the term bile salts to denote bile components having steroid nuclei with free acidic groups, including taurocholic and glycocholic acids.
- Abbreviations:
- TFA
- trifluoroacetic acid
- CHO
- glucose or maltose
- GITC
- guanidylisothiocyanate
- HPLC
- high pressure liquid chromatography
- Received May 22, 1996.
- Accepted September 9, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}