


Abstract
The antihypertensive agent diltiazem (DTZ) impairs hepatic drug metabolism by inhibition of cytochrome P450 (CYP). The accumulation of DTZ metabolites in serum occurs during prolonged therapy and leads to decreased DTZ elimination. Thus, DTZ metabolites may contribute to CYP inhibition. This study assessed the role of human CYPs in microsomal DTZ oxidation and the capacity of DTZ metabolites to inhibit specific CYP activities. DTZ N-demethylation varied 10-fold in microsomal fractions from 17 livers (0.33–3.31 nmol/mg of protein/min). DTZ oxidation was correlated with testosterone 6β-hydroxylation (r = 0.82) and, to a lesser extent, tolbutamide hydroxylation (r = 0.59) but not with activities mediated by CYP1A2 or CYP2E1. CYP3A4 in lymphoblastoid cell microsomes catalyzed DTZ N-demethylation but CYP2C8 and CYP2C9 were also active (∼20% and 10% of the activity supported by CYP3A4); seven other CYPs produced little or no N-desmethyl DTZ from DTZ. The CYP3A4 inhibitors ketoconazole and troleandomycin decreased microsomal DTZ oxidation, but inhibitors or substrates of CYP2C, CYP2D and CYP2E1 produced no inhibition. Some inhibition was produced by α-naphthoflavone, a chemical that inhibits CYP1As and also interacts with CYP3A4. In further experiments, the capacities of DTZ and three metabolites to modulate human CYP 1A2, 2E1, 2C9 and 3A4 activities were evaluated in vitro. DTZ and its N-desmethyl and N,N-didesmethyl metabolites selectively inhibited CYP3A4 activity, whereas O-desmethyl DTZ was not inhibitory. The IC50 value of DTZ against CYP3A4-mediated testosterone 6β-hydroxylation (substrate concentration, 50 μM) was 120 μM. The N-desmethyl (IC50 = 11 μM) and N,N-didesmethyl (IC50 = 0.6 μM) metabolites were 11 and 200 times, respectively, more potent. From kinetic studies, N-desmethyl DTZ and N,N-didesmethyl DTZ were potent competitive inhibitors of CYP3A4 (Ki = ∼2 and 0.1 μM, respectively). CYP3A4 inhibition was enhanced when DTZ and N-desmethyl DTZ underwent biotransformation in NADPH-supplemented hepatic microsomes in vitro, supporting the contention that inhibitory metabolites may be generated in situ. These findings suggest that N-demethylated metabolites of DTZ may contribute to CYP3A4 inhibition in vivo, especially under conditions in which N-desmethyl DTZ accumulates, such as during prolonged DTZ therapy.
The calcium channel antagonist DTZ has been shown to inhibit mammalian CYPs (Renton, 1985) and to precipitate pharmacokinetic interactions with drugs such as carbamazepine (Brodie and Macphee, 1986), cyclosporin A (Combalertet al., 1989) and nifedipine (Toyosaki et al., 1988). Because CYP3A4 participates in the oxidation of these drugs, it seems likely that this enzyme may be a target for inhibition by DTZ. Although there is evidence from animal studies that CYP3As may be involved in DTZ metabolism, the contribution of CYPs to the reaction in human liver has not been evaluated. Furthermore, the selectivity of DTZ as an inhibitor of major CYP reactions in human liver remains to be established.
Apart from the inhibitory effects of DTZ on drug oxidation, evidence from the literature shows that DTZ metabolites accumulate during prolonged therapy and may contribute to CYP inhibition (Abernethy and Montamat, 1987; Montamat and Abernethy, 1987). In this study, we also investigated the capacity of DTZ and its oxidized metabolites (fig.1) to inhibit the activities of four major human CYPs and determined the role of hepatic biotransformation in the generation of DTZ metabolites with greater inhibitory potency toward CYP enzymes.

Structure of DTZ and metabolites. In DTZ R1 = N(CH3)2, R2 = CH3; in N-desmethylDTZ R1 = NHCH3, R2 = CH3; in N,N-didesmethylDTZ R1= NH2, R2 = CH3; in O-desmethylDTZ R1 = N(CH3)2, R2 = H.
The principal findings to emerge from the present study were that DTZ N-demethylation is catalyzed extensively by CYP3A4, probably the major drug oxidase in human liver, and that DTZ is also a preferential inhibitor of CYP3A4 activity; DTZ did not inhibit reactions mediated by CYPs 1A2, 2C9 or 2E1. Importantly, both N-desmethyl and N,N-didesmethyl DTZ were significantly more potent than DTZ as inhibitors of CYP3A4. Kinetic studies indicated that the two metabolites elicited inhibition of CYP3A4 activity by a competitive mechanism. In vitrobiotransformation of DTZ in human microsomes led to an increase in the observed extent of inhibition of CYP3A4 activity, as did the incubation of N-desmethyl DTZ with NADPH-fortified microsomes. Taken together, these results suggest that CYP3A4 contributes extensively to DTZ N-demethylation in human liver and that the N-demethylated metabolites produced may accumulate during chronic DTZ therapy and lead to inhibition of the enzyme.
Materials and Methods
Chemicals.
DTZ HCl (d-cis-isomer), 7-ethylresorufin (also termed resorufin-7-ethyl ether or 7-ethoxyresorufin), tolbutamide, N-nitrosodimethylamine, 7-ethoxycoumarin and biochemicals were purchased from Sigma Aldrich (Castle Hill, NSW, Australia). DTZ metabolites were gifts from Tanabe Seiyaku Company (Osaka, Japan). [4-14C]Testosterone (specific activity, 57–59 mCi/mmol), Hyperfilm-MP and scintillation fluid (ACS-II) were purchased from Amersham Australia (North Ryde, NSW, Australia). Steroid metabolites were obtained from Steraloids (Wilton, NH) or the Medical Research Council Steroid Reference Collection (Queen Mary’s College, London, UK). Aniline was obtained from Ajax Chemicals (Sydney, Australia) and redistilled from zinc dust before use. Analytical reagent and HPLC grade solvents and other chemicals were from Ajax or Rhone-Poulenc (Sydney, Australia). Silica gel 60 F254 plates for TLC were from Alltech (Sydney, Australia).
Liver donors and preparation of microsomal fractions.
Ethical approval for experiments on human tissue was granted by the Human Ethics committee of the Western Sydney Area Health Service. Tissue was obtained under the organ donation scheme from accident victims, through either the Queensland or Australian Liver Transplant Programs (Princess Alexandria Hospital, Brisbane, Queensland, and Royal Prince Alfred Hospital, Sydney, NSW, Australia, respectively). In most cases, samples were excess tissue from adult donors used in the transplantation of pediatric recipients, but several samples were from the normal margin of tissue taken for biopsy during liver resection. Tissue was obtained from the operating theater, perfused with ice-cold ViaSpan solution (Belzer University of Wisconsin solution; DuPont, Wilmington, DE), packed in ice and transported to the laboratory where it was snap-frozen in liquid nitrogen. Washed microsomes were prepared by centrifugation and were resuspended in 50 mM potassium phosphate buffer, pH 7.4, containing 1 mM EDTA and 20% glycerol for storage at −70°C (Murray et al., 1986). Seventeen individual livers were used in this study (table 1). Microsomal protein contents were determined according to the Lowry procedure (Lowryet al., 1951); bovine serum albumin was used as the standard.
Interindividual variation in DTZ N-demethylation in human hepatic microsomes
Cell microsomes containing cDNA-derived human CYPs.
Microsomes from human lymphoblastoid cell lines (AHH-1 TK+/−) in which cDNA-derived human CYPs (1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4 and 4A11) had been expressed were purchased from Gentest Corp. (Woburn, MA). Control microsomes were prepared from untransfected AHH-1 TK+/− cells. Protein contents of the preparations were 10 mg/ml; CYP contents were determined by the supplier (table 2).
In vitro oxidation of DTZ by cDNA-derived human P450s
Assays of human microsomal CYP activities.
Testosterone hydroxylation (0.15 mg of protein/0.4 ml incubation;14C-testosterone 50 μM, 0.18 μCi) was measured as previously described (Murray, 1992). Reactions were conducted in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 2.5 min. Radioactive products were extracted from incubations, separated by TLC, located by autoradiography and quantified according to methods outlined previously (Murray, 1992).
For IC50 values, rates of steroid hydroxylation were determined in duplicate in each of three individual human hepatic microsomal fractions using five concentrations of inhibitor. IC50 values were calculated from plots of percentage activity remaining vs. log10 inhibitor concentration. In kinetic experiments, testosterone concentrations over the range 10 to 200 μM were used. Similarly, concentrations of inhibitors were varied over the ranges of 0 to 3 μM (N,N-didesmethyl DTZ) and 0 to 60 μM (N-desmethyl DTZ), respectively. Data that were derived were subjected to graphical analyses outlined in Segel (1975). Briefly, initial Lineweaver-Burk (1/V vs. 1/S) and Dixon (1/S vs. I) analyses were followed by the construction of primary replots (slopes of lines in the Lineweaver-Burk and Dixon plots as a function of I or 1/S, respectively). The appearance of the replots can be used to characterize the nature of the inhibition, andKi values are derived from the same analysis (Segel, 1975).
Aniline 4-hydroxylation in human liver microsomes (1.6 mg of protein/0.6 ml incubation; substrate concentration 5 mM) was measured as described previously (Murray and Ryan, 1982). Reactions were conducted in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 12 min. 4-Aminophenol formation was detected by the formation of a blue complex with alkaline phenol and quantified at 610 nm.
N,N-Dimethylnitrosamine N-demethylation in human hepatic microsomes (2.5 mg of protein/1.0 ml incubation; substrate concentration 4 mM) was measured as described by Peng et al. (1983). Reactions were conducted in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 20 min.
7-Ethylresorufin O-deethylation activity (0.5 mg of protein/2 ml incubation, substrate concentration 2.5 μM) was measured in Tris · HCl buffer (0.1 M, pH 7.8) using the continuous spectrofluorometric procedure of Prough et al. (1978). Resorufin formation was monitored at the excitation/emission wavelength pair of 560/580 nm.
Tolbutamide 4-hydroxylation activity (0.3 mg of protein/0.4 ml incubation, tolbutamide 300 μM) was measured using the method ofKnodell et al. (1987). Reactions were conducted in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 15 min. After the addition of HCl, the products were extracted with diethyl ether and the residue was evaporated, reconstituted in acetonitrile and applied to a Beckman Ultrasphere C18 column (5 μm, 25 cm × 4.6 mm i.d., Beckman Instruments Inc., San Ramon, CA). The mobile phase was 0.05% phosphoric acid, pH 2.6/acetonitrile (3:2). Retention times were 4.14 min for 4-hydroxytolbutamide, 9.25 min for chlorpropamide (internal standard) and 11.53 min for tolbutamide.
Oxidation of DTZ in human hepatic and lymphoblastoid cell microsomes.
DTZ oxidation was determined in human hepatic microsomes at a substrate concentration of 100 μM, except in the determination of kinetic parameters, where the substrate range was 5 to 200 μM. Incubations (0.15 mg of protein/400 μl) were conducted in potassium phosphate buffer (0.1 M, pH 7.4) containing EDTA (1 mM) at 37°C for 10 min. Preliminary studies established that product formation was linear over the time indicated. Reactions were terminated by freezing in an acetone/dry ice bath. After subsequent thawing, the internal standard was added (imipramine, 30 nmol), and the solution was basified by the addition of 1 g of NaHCO3 and then extracted with ethyl acetate (4 ml). The organic phase (3.8 ml) was extracted with HCl (0.01 M, 200 μl), which was then evaporated under a stream of nitrogen and dissolved in 200 μl of 2 mM HCl (Hussainet al., 1992) for separation by HPLC on an Ultrasphere C18 column (5 μm, 25 cm × 4.6 mm i.d.). The mobile phase was 0.05 M KH2PO4 (pH 2.9), acetonitrile and triethylamine (60:38.8:0.2), the flow rate was 1.5 ml/min and the detection wavelength was 238 nm (Ascalone et al., 1994). Retention times were 3.1 min for N,N-didesmethyl DTZ and O-desmethyl DTZ, 3.6 min for N-desmethyl DTZ, 5.1 min for deacetyl DTZ, 7.8 min for DTZ and 11.6 min for imipramine (fig. 2).

Typical HPLC elution profile of DTZ and metabolites formed in human hepatic microsomal incubations (imipramine is the internal standard).
DTZ oxidation supported by cDNA-derived CYPs in lymphoblastoid cell microsomes was also determined. The supplier’s assay instructions were followed. Briefly, DTZ (100 μM) was incubated with NADPH (1 mM) and cell microsomes (at a protein concentration of 1 mg/ml) over a 60-min period. Reactions were terminated essentially as described for hepatic microsomal reactions, and the products were separated by HPLC. 7-Ethoxycoumarin O-deethylation activities of the preparations were also determined (Prough et al., 1978) because this activity is known to be catalyzed by a number of individual CYPs (Chang et al., 1993). Reactions contained 0.4 mg of microsomal protein and 400 μM ethoxycoumarin.
In further experiments to assess the effect of DTZ or N-desmethyl DTZ biotransformation on CYP3A4 activity, these agents were incubated with NADPH-fortified microsomes before transfer to tubes containing14C-testosterone. Reactions were then continued for the usual 2.5 min, after which radioactive metabolites were extracted and separated as before.
Data analysis and statistics.
Values are presented throughout as mean ± S.E.M. of triplicate estimates; except where stated otherwise. Differences between means of several treatments were detected using analysis of variance and Dunnett’s q′-test.
Results
Interindividual variation in DTZ oxidation in human liver.
Microsomal fractions from 17 individual human livers were assessed in this study. Where available, the recent drug histories of the donors are also indicated in table 1. Three of the individuals were cigarette smokers. As shown in table 1, the individual variation in DTZ (100 μM) oxidation to its major metabolite N-desmethyl DTZ was considerable (range, 0.33–3.31 nmol/mg of protein/min;n = 17).
A kinetic analysis of DTZ N-demethylation was undertaken in three human hepatic fractions (HL27, HL30 and HL31). From Hanes-Woolf plots, the Michaelis constant (Km ) for the N-demethylation reaction was calculated to be 23 ± 10 μM. Maximal reaction velocities (V max) for the reactions were also determined and found to be 5.36, 1.65 and 0.99 nmol/mg of protein/min for HL27, HL30 and HL31, respectively. The Hanes-Woolf plot for DTZ N-demethylation carried out in HL31 is shown in figure3.

Representative Hanes-Woolf plot of DTZ N-demethylation in human hepatic microsomes.
Correlation of DTZ N-demethylation with other microsomal oxidations.
As shown in table 3, in microsomal fractions from 17 individual livers, rates of DTZ N-demethylation were well correlated with CYP3A4-mediated testosterone 6β-hydroxylation and, to a lesser extent, CYP2C-mediated tolbutamide hydroxylation (r = 0.82; P < .001; r = 0.59; P < .05, respectively, fig. 4). In contrast, correlations between DTZ oxidation and aniline 4-hydroxylation, 7-ethylresorufin O-deethylation and N,N-dimethylnitrosamine N-demethylation did not attain statistical significance. In this subset of 17 microsomal fractions, testosterone 6β-hydroxylation (CYP3A4) and tolbutamide hydroxylation (CYP2C) activities were correlated (r = 0.61; P < .01), as were the hydroxylations of tolbutamide and aniline (CYP2C and CYP2E1, respectively; r = 0.65, P < .05,n = 14) and the oxidations of aniline and N,N-dimethylnitrosamine N-demethylation; correlations between other pairs of substrate oxidation activities were not significant. Due to limitations of sample availability, aniline 4-hydroxylation and N,N-dimethylnitrosamine N-demethylation activities were determined on only 14 of the microsomal fractions.
Correlation matrix between DTZ N-demethylation and other microsomal oxidations mediated by human CYPs

Correlation of DTZ N-demethylation with (A) testosterone 6β-hydroxylation and (B) tolbutamide hydroxylation in human hepatic microsomes.
Modulation of DTZ oxidation in human liver by chemical agents.
The modulatory effects of chemical substrates or inhibitors of specific human CYP enzymes on DTZ N-demethylation were evaluated. It is evident from the data in figure 5 that the most pronounced effects were produced by the inhibitors of CYP3A4. Thus, ketoconazole (25 μM) and troleandomycin (500 μM) decreased DTZ oxidation to ∼10% of control activity; an IC50 value of 0.13 μM was determined for ketoconazole. α-Naphthoflavone, generally considered to be a CYP1A2 inhibitor but also known to interact as an activator of CYP3A4, decreased DTZ oxidation activity in microsomal fractions from three individual livers to ∼50% of control when incorporated into incubations at a concentration of 100 μM (fig. 5). Tolbutamide (300 μM; 2C8/9 substrate), sulfaphenazole (50 μM; 2C9 inhibitor), debrisoquine sulfate (100 μM; 2D6 substrate) and acetaminophen (1 mM; substrate for 1A2 and 2E1) were essentially without effect on microsomal DTZ N-demethylation.
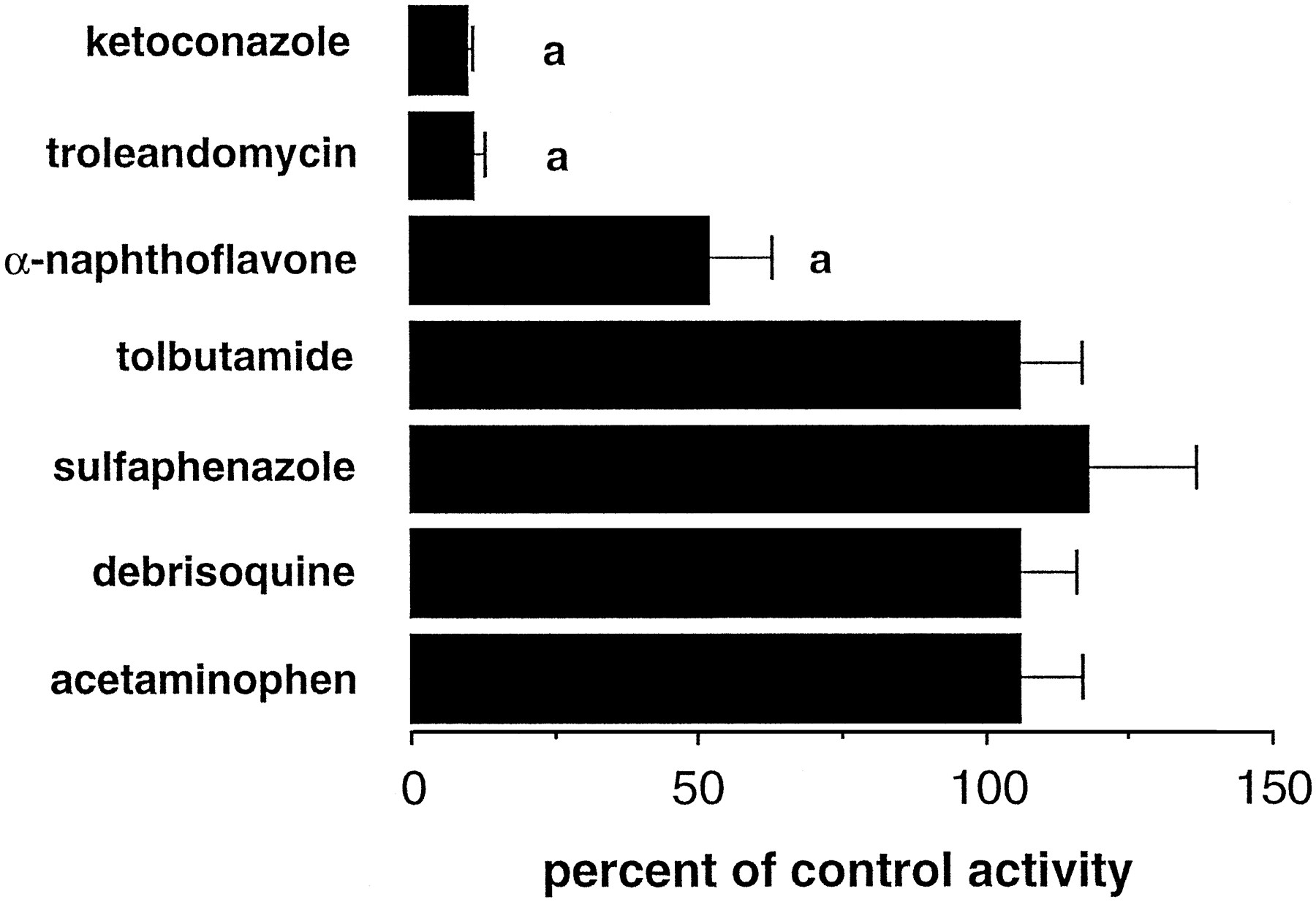
Effect of CYP-specific inhibitors and substrates on microsomal DTZ N-demethylation in human liver. Results are presented as the mean ± S.E.M. of determinations in three individual human liver preparations. aSignificant difference from control (P < .01).
N-Demethylation of DTZ by cDNA-derived human CYPs.
To investigate further the participation of human CYPs in DTZ N-demethylation, the capacities of cDNA-derived CYPs to support the reaction in lymphoblastoid cell microsomes were assessed (100 μM). Thus, CYPs 3A4, 2C8 and 2C9 catalyzed DTZ oxidation to N-desmethyl DTZ. CYP3A4 produced 9.92 pmol of product/hr/pmol of CYP, whereas 2C8 and 2C9 generated the metabolite at ∼20% and ∼10%, respectively, of the rate exhibited by 3A4. CYPs 1A1, 1A2, 2A6, 2B6, 2D6, 2E1 and 4A11 exhibited little or no activity in DTZ N-demethylation (table 2). 7-Ethoxycoumarin O-deethylation activities catalyzed by nine of the preparations were determined (range, 0.25–32 pmol/hr/pmol of P450) because this reaction is mediated by a number of CYPs and can be used for comparative purposes (Chang et al., 1993).
Inhibition of CYP3A4-dependent steroid 6β-hydroxylation by DTZ and its metabolites.
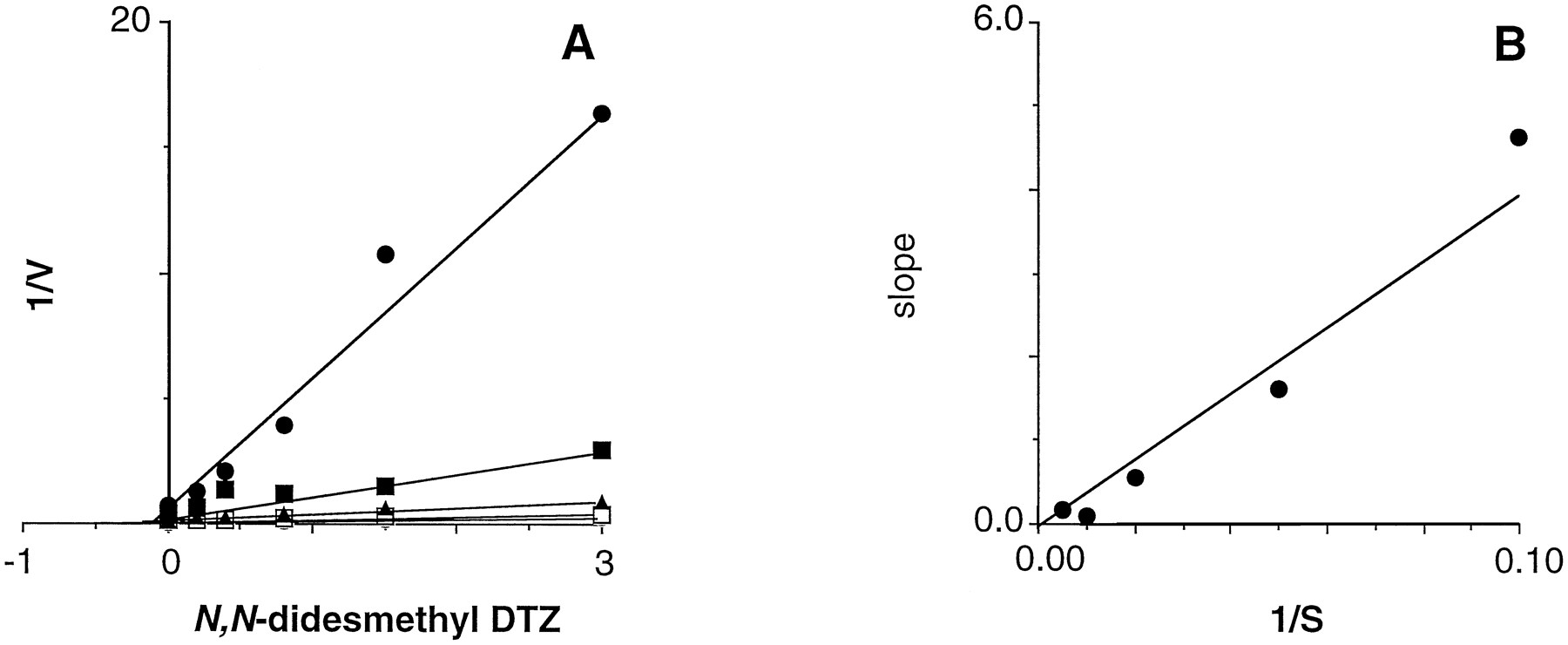
Of the four compounds tested, three were found to be inhibitory. The parent drug DTZ exhibited an IC50 value of 120 μM in microsomes (data obtained with three separate fractions). N-Desmethyl DTZ and N,N-didesmethyl DTZ were, respectively, 11- and 200-fold more potent (IC50values of 11 and 0.6 μM) than DTZ as an inhibitor of CYP3A4 activity; the O-demethylated metabolite of DTZ did not inhibit the activity. The mechanism of inhibition of CYP3A4 activity by these two metabolites was assessed in kinetic experiments. As shown in figures 6and 7, both N-desmethyl DTZ and N,N-didesmethyl DTZ were competitive inhibitors of CYP3A4-mediated testosterone 6β-hydroxylation. Thus, Ki values for N-desmethyl DTZ were 2.2 and 2.5 μM in microsomes from HL26 and HL27, respectively, and the corresponding values for N,N-didesmethyl DTZ were 0.07 and 0.10 μM, respectively).
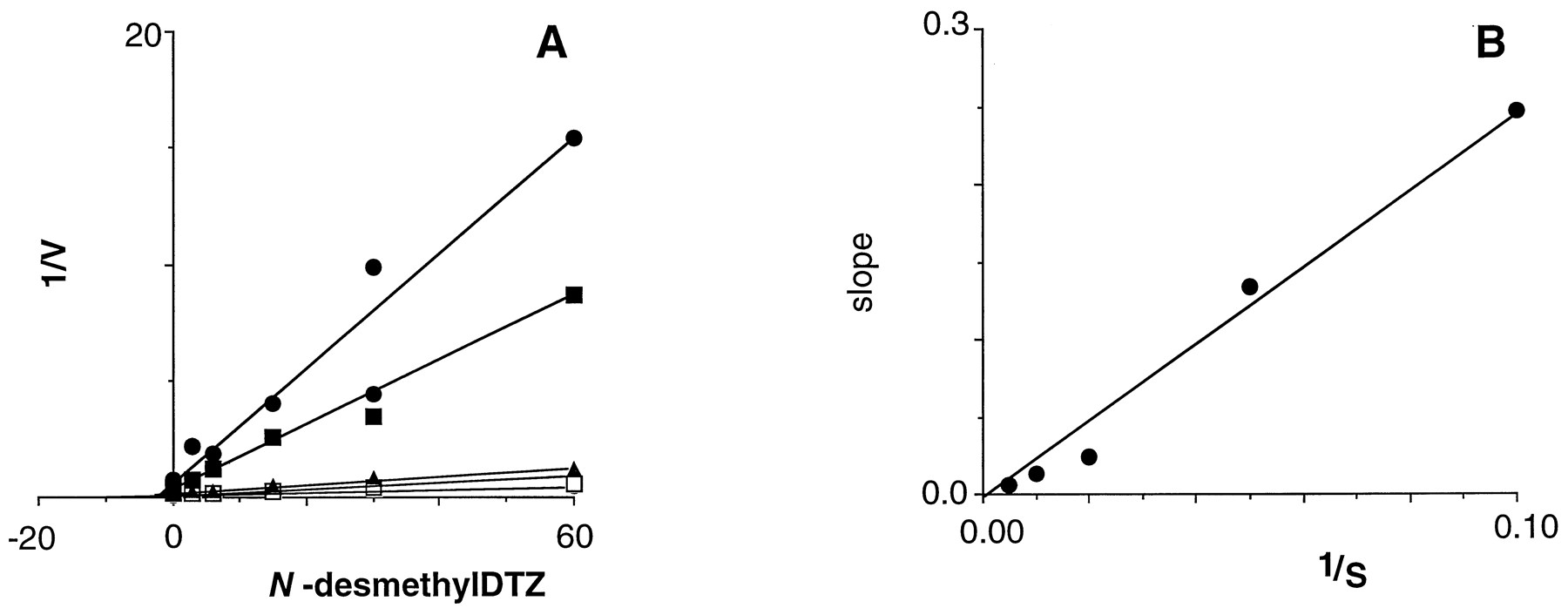
Kinetics of the inhibition of microsomal CYP3A4-mediated testosterone 6β-hydroxylation in human liver by N-desmethyl DTZ. A, Dixon plots at several testosterone concentrations: •, 10 μM; ▪, 20 μM; ▴, 50 μM; ○, 100 μM; □, 200 μM. B, Dixon plot slopes vs. reciprocal testosterone concentrations. Units are substrate and inhibitor concentrations, μM; V, nmol/min/mg of protein.

Kinetics of the inhibition of microsomal CYP3A4-mediated testosterone 6β-hydroxylation in human liver N,N-desmethyl DTZ. A, Dixon plots. B, Dixon replots. See legend to figure 6 for other details.
7-Ethylresorufin O-deethylation (catalyzed by CYP1A2) was essentially refractory to inhibition by DTZ and its metabolites. No inhibition of the activity was observed in three individual livers, although 40% inhibition was produced by 200 μM N,N-didesmethyl DTZ in HL24. Similar findings were made in the cases of tolbutamide methyl hydroxylation and aniline 4-hydroxylation (2C9 and 2E1 mediated, respectively). No inhibition of either activity was noted when DTZ or metabolites were tested at concentrations up to 200 μM.
Potentiation of CYP3A4 inhibition during DTZ and N-desmethyl DTZ biotransformation in human hepatic microsomes.
From the foregoing experiments it was apparent that DTZ underwent CYP-mediated oxidation to N-desmethyl DTZ and that this metabolite is an effective inhibitor of CYP3A4 activity. Subsequent studies assessed the effect of DTZ metabolism on the extent of CYP3A4 inhibition in microsomal incubations. Thus, at a concentration of 100 μM, with preincubation times up to 17.5 min, NADPH-supported microsomal DTZ biotransformation enhanced the extent of inhibition of testosterone 6β-hydroxylation (fig. 8). Under such conditions, the concentration of N-desmethyl DTZ formed during the course of the incubation was ∼6.5 μM. Therefore, because N-desmethyl DTZ was an effective inhibitor of testosterone 6β-hydroxylation (IC50 = 12 μM), it is feasible that this metabolite was responsible for the enhanced inhibition of CYP3A4 activity.

Time-dependent effects of preincubation of (•) DTZ or (▪) N-desmethyl DTZ with NADPH-supplemented human hepatic microsomes on CYP3A4-mediated testosterone 6β-hydroxylation. The agents (25 μM) were preincubated with NADPH and human hepatic microsomes at 37°C for varying periods before transfer to vials containing testosterone. Reactions then continued for 2.5 min, after which reactions were terminated and product formation was estimated. Note the logarithmic scale (base 10) on the y-axis.
N,N-Didesmethyl DTZ was substantially more potent as an inhibitor of CYP3A4 activity. To assess whether this metabolite may contribute to overall CYP3A4 inhibition, N-desmethyl DTZ oxidation was determined in NADPH-supplemented microsomal fractions; metabolism to N,N-didesmethyl DTZ was slow but detectable (0.011 ± 0.002 nmol/mg of protein/min). Again, during microsomal N-desmethyl DTZ oxidation, the inhibition of CYP3A4-dependent testosterone 6β-hydroxylation was enhanced.
Discussion
The results of the present study implicate CYP3A4, and perhaps other members of the CYP3A subfamily, as the major enzyme active in DTZ N-demethylation in human liver. The supporting evidence can be summarized as follows: (1) the correlation between rates of CYP3A4-dependent testosterone 6β-hydroxylation and DTZ N-demethylation was highly significant, (2) the CYP3A4 inhibitors ketoconazole and troleandomycin inhibited DTZ oxidation potently and (3) of the 10 cDNA-derived CYPs in lymphoblastoid cell microsomes that were evaluated, CYP3A4 was the most active catalyst of DTZ N-demethylation. A good correlation between CYP2C9-mediated tolbutamide hydroxylation and DTZ N-demethylation was also observed in human hepatic microsomes (although the correlation between testosterone 6β-hydroxylation and tolbutamide hydroxylation was significant in the subset of livers used in the present investigation). Notwithstanding this point, cDNA-derived CYPs 2C8 and 2C9 supported DTZ oxidation in lymphoblastoid cell microsomes. Thus, there is some evidence that CYPs from the 2C subfamily may participate in the oxidation of DTZ in liver, but they may be minor catalysts of the reaction.
CYP3A protein is quantitatively significant in human liver.Shimada et al. (1994) documented that 30% to 50% of total CYP in human hepatic microsomes was immunoreactive with an anti-CYP3A IgG. It is also apparent from the recent literature that CYP3A4 is involved in the enzymatic oxidation of an increasing list of drugs, including midazolam (Kronbach et al., 1989), alfentanil (Yunet al., 1992), lansoprazole (Pichard et al., 1995) and docetaxel (Marre et al., 1996). CYP3A4 also activates carcinogens and mutagens, like aflatoxin B1 and sterigmatocystin (Shimada and Guengerich, 1989; Shimada et al., 1989), to the toxic species. Certainly, impaired elimination of drugs that are metabolized extensively by CYP3A4, resulting in their accumulation in serum could lead to adverse effects, including enhanced therapeutic effect.
The potential for DTZ to inhibit CYP-mediated drug oxidation has been known for some time, and clinically significant pharmacokinetic interactions have been reported during concurrent drug therapy (Brodie and Macphee, 1986). However, the specificity of the interaction of DTZ with human CYPs has not been completely characterized. One of the major findings from the present study is that DTZ is a selective inhibitor of CYP3A4, which is involved in the enzymic oxidation of many therapeutic agents. Thus, it was found that the 6β-hydroxylation of testosterone in human hepatic microsomes, a reaction characteristic of CYP3A4 (Waxman et al., 1988), was inhibited by DTZ (IC50 = 120 μM). The relative affinity of CYP3A4 for DTZ and testosterone is reflected by the respectiveKm values of 23 μM (present study; fig. 3) and 55 μM (present study; data not shown). In a previous study, we determined a Km value of 94 μM for testosterone 6β-hydroxylation in human liver (Murray et al., 1994). These data suggest that testosterone has a somewhat lower affinity than DTZ for CYP3A4 and therefore are consistent with the inhibition of CYP3A4 that was observed in the present study. In contrast, 7-ethylresorufin O-deethylation, catalyzed by CYP1A2 (Tassaneeyakul et al., 1993); tolbutamide 4-hydroxylation, catalyzed by CYP2C9 (Knodell et al., 1987); and aniline 4-hydroxylation, catalyzed principally by CYP2E1 (Wrighton and Stevens, 1992), were refractory to inhibition by DTZ.
The administration of DTZ by multiple dosage regimen leads to a prolongation of the half-life of DTZ (Montamat and Abernethy, 1987). Accordingly, the present study also evaluated the role of DTZ metabolites in CYP inhibition. Of the three DTZ metabolites examined, N-desmethyl and N,N-didesmethyl DTZ were 11- and 200-fold, respectively, more potent than DTZ as inhibitors of CYP3A4; O-desmethyl DTZ was not inhibitory. The selectivity of inhibition by these metabolites was similar to that of the parent compound. Thus, neither N-desmethyl nor N,N-didesmethyl DTZ inhibited activities associated with CYPs 1A2, 2E1 or 2C9. The mechanism by which the two metabolites modulate CYP3A4 activity was explored in the present study using a kinetic approach. It was evident that both metabolites were competitive inhibitors of CYP3A4 activity: N-desmethyl exhibited aKi value of ∼2 μM, and the N,N-didesmethyl analog exhibited a Ki value of ∼0.1 μM. Thus, it is clear that both metabolites, especially the N,N-didesmethyl compound, have high capacity to interact with the enzyme. Indeed, from a comparison of these Ki values with theKm value for testosterone 6β-hydroxylation, it appears that CYP3A4 has ∼25- and ∼500-fold greater affinity for the N-desmethyl and N,N-didesmethyl metabolites than for the substrate. Thus, these in vitro kinetic data provide a basis for interpretation of the considerable capacity of the metabolites to inhibit CYP3A4 activity in vivo. It should be borne in mind that the inhibition of CYP3A4 by DTZ metabolites, although quite potent, is reversible and should be distinguished from processes such as metabolite-intermediate complexation and mechanism-based inactivation. Inhibition processes of that type are characterized by the generation of destructive metabolites that can produce long-term inhibition of CYP enzymes (Murray and Reidy, 1990). However, the formation of drug metabolites that are potent reversible CYP inhibitors may be a more widespread phenomenon. This possibility requires continuing evaluation in further studies.
Preincubation of DTZ or N-desmethyl DTZ with NADPH-fortified microsomes significantly increased the extent of inhibition of testosterone 6β-hydroxylation. N-Desmethyl DTZ concentrations of ∼6.5 μM were detected and appeared to be increasing in a linear fashion (not shown); such concentrations of N-desmethyl DTZ are sufficient to produce substantial inhibition of CYP3A4. The more potent N,N-didesmethyl metabolite was also detected in incubations containing N-desmethyl DTZ. It is therefore possible that the N,N-didesmethyl DTZ detected in serum in previous studies (Sugawara et al., 1988) may be formed in liver from its precursor N-desmethyl DTZ.
In summary, this study has found that of the four major human CYPs, only CYP3A4 is inhibited by DTZ and its major hepatic metabolitesin vitro. The other major CYPs, 1A2, 2E1 and 2C9, are not affected by DTZ, so pharmacokinetic interactions between DTZ and drugs metabolized by these CYPs are unlikely. In vitrobiotransformation of DTZ by CYP3A4 produces N-demethylated metabolites that are potent inhibitors of the activity of the enzyme. These metabolites are likely to be responsible for the impaired elimination of DTZ under multiple dosing regimen in vivo.
Acknowledgments
The assistance of the Australian and Queensland Liver Transplant Programs in obtaining human liver is gratefully acknowledged. We thank Tanabe Seiyaku Co. (Osaka, Japan) for their generous gifts of authentic DTZ metabolites and Dr. H. Smith (ICI, Australia) for assistance in obtaining these compounds.
Footnotes
-
Send reprint requests to: Dr. Michael Murray, Department of Medicine, Westmead Hospital, Westmead, NSW 2145, Australia.
-
↵1 This work was supported by a grant from the National Health and Medical Research Council of Australia.
- Abbreviations:
- DTZ
- diltiazem
- HPLC
- high pressure liquid chromatography
- CYP
- cytochrome P450
- TLC
- thin-layer chromatography
- Received November 15, 1996.
- Accepted March 20, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}