


Abstract
The 4′-hydroxylation of the S-enantiomer of the anticonvulsant drug mephenytoin exhibits a genetic polymorphism in humans. This polymorphism shows marked interracial heterogeneity, with the poor metabolizer (PM) phenotype representing 2 to 5% of Caucasian and 13 to 23% of Asian populations. Two defective CYP2C19alleles, CYP2C19*2 and CYP2C19*3, have been described which account for ∼87% of Caucasian and >99% of Oriental PM alleles. The present study identifies a new allele (CYP2C19*4) in Caucasian PMs which contains an A → G mutation in the initiation codon. A new polymerase chain reaction-restriction fragment length polymorphism genotyping test was developed, and the incidence of this allele was examined in a European Caucasian population which had been phenotyped for mephenytoin metabolism. One of nine putative PMs was heterozygous forCYP2C19*2/CYP2C19*4, which suggests thatCYP2C19*4 represents a defective allele. Six of the seven remaining putative PMs available for genotyping were explained byCYP2C19*2. The frequency of the CYP2C19*4 allele in Caucasians was 0.6%. An additional Caucasian PM from a separate study was also heterozygous for CYP2C19*2 andCYP2C19*4. To verify that CYP2C19*4 represented a defective CYP2C19 allele, the initiation codon of the normal CYP2C19*1 cDNA was mutated to a GTG, and both cDNAs were expressed in yeast. Recombinant CYP2C19 protein was detected by Western blot analysis of colonies transformed with CYP2C19*1 cDNA, but not in those transformed with CYP2C19*4 cDNA. The two cDNAs were also used in an in vitro coupled transcription/translation assay. CYP2C19 protein was translated only from the CYP2C19*1 allele. These data indicate that CYP2C19*4 represents a new PM allele.
A well-characterized genetic polymorphism of drug oxidation in humans is associated with impaired 4-hydroxylation of the S-enantiomer of the anticonvulsant drug mephenytoin in humans (Wilkinson et al., 1989). Individuals can be characterized phenotypically as EMs or PMs. This polymorphism also affects the metabolism of several other clinically used drugs such as the anti-ulcer drug omeprazole (Anderssonet al., 1992), certain barbiturates (Küpfer and Branch, 1985, Adedoyin et al., 1994) and antidepressants (Sindrup et al., 993, Baumann et al., 1986,Nielsen et al., 1994, Skjelbo et al., 1991), the antimalarial proguanil (Ward et al., 1991), propranolol (Ward et al., 1989) and diazepam (Bertilsson et al., 1989). The incidence of the PM trait is much more common (13–23%) in Oriental populations than in Caucasians (2–5%) (Wilkinson et al., 1989). CYP2C19 is the enzyme responsible for this polymorphism (Wrighton et al., 1993, Goldsteinet al., 1994). Our laboratory has previously identified two genetic defects in CYP2C19, originally termedCYP2C19m1 and CYP2C19m2, but more recently termed CYP2C19*2 and CYPC19*3 by a new nomenclature system proposed by Daly et al.(1996),4 which account for >99% of Oriental but only ∼87% of Caucasian PM alleles (de Morais et al.,1994a, b). Therefore, it seems likely that additional defective alleles contribute to the PM phenotype in Caucasians. The present study identifies an additional defective CYP2C19 allele in Caucasian PMs of mephenytoin.
Materials and Methods
Subjects.
An American Caucasian EM subject (CE) and a Canadian Caucasian PM subject (CB) were patients about whom information was obtained from previously reported studies (de Morais et al., 1994a, b; Sarich et al., 1997). A European Caucasian population consisted of the control group (234 unrelated individuals of which 219 were males and 15 were females) from a case-control study of tobacco-related cancers in smokers (Benhamouet al., 1997). These individuals were smokers without previous or current malignant disease recruited from 10 French private or public hospitals and were predominantly male.
Phenotyping.
The American and Canadian subjects ingested 100 mg racemic mephenytoin after emptying their bladders and a 0- to 8-hr urine sample was collected. The PM phenotype was defined as a urinary mephenytoin S/R ratio of greater than 0.9, and recovery of less than 1% of the dose in the urine as 4′-hydroxymephenytoin (Wedlund et al., 1984, Sarich et al., 1997). S/R ratios were determined both before and after acidification of the urine to circumvent misclassification because of the presence of an acid-labile metabolite in some urines (Wedlund et al., 1987; Zhanget al., 1992).
The European population was given a capsule containing 100 mg racemic mephenytoin and 25 mg dextromethorphan hydrobromide after emptying their bladders, and a 8- to 12-hr urine sample was collected. Metabolism of these drugs is mediated by two distinct P450 enzymes (CYP2C19 and CYP2D6), and they can be coadministered without any interaction (Guttendorf et al., 1990). The amount of 4′-hydroxymephenytoin in the urine was measured by reversed phase high-performance liquid chromatography adapted from the method of Meieret al. (1985). The HI represents the molar ratio ofS-mephenytoin administered to that of 4′-hydroxymephenytoin recovered in the urine (Küpfer and Preisig, 1984). S/R ratios before and after acidification of the urine were also determined on samples with HI values >18 before assigning phenotypes.
CYP2C19 genotyping and sequencing ofCYP2C19*4.
Genomic DNA was isolated from blood as described previously (Sambrook et al., 1989) or using Qiamp blood kits (Qiagen, Chatsworth, CA) according to the manufacturer’s protocol. To identify new genetic defects of CYP2C19, exons and exon-intron junctions from various subjects were amplified by PCR with intron-specific primers, and the PCR products were sequenced on an automated sequencer (Applied Biosystems, Foster City, CA) with the PRISM Dye Terminator Cycle Sequencing kit. Genotyping procedures for detection of the CYP2C19*2 and CYP2C19*3 alleles have been described previously (Goldstein and Blaisdell, 1996).
A mismatch PCR-RFLP genotyping test was developed for the CYP2C19*4 allele. This test used a forward PCR primer from the 5′-upstream region with a 1-bp mismatch (underlined) and a 6-bp A and T overhang on the 5′-end (underlined) (5′-ATTATATTAACAAGAGGAGAAGGCTGCA-3′) and a 2C19-specific reverse primer (5′-TTGGTTAAGGATTTGCTGACA-3′) from exon 1 in a method similar to that described by Wang et al. (1995)for CYP2C9 alleles. The mismatched primer introduced aPstI site only in the CYP2C19*4 allele. The amplification procedure was similar to that described for the detection of CYP2C19*2 and CYP2C19*3 (Goldstein and Blaisdell, 1996) except that the annealing temperature was 57°C for 15 sec, extension time 15 sec and the number of cycles was 40. The resulting 195-bp products were digested with 4 units of PstI at 37°C for 2 hr and the fragments were separated on 4% agarose gels. Samples containing the CYP2C19*4 alleles produced 167-bp and 28-bp fragments after PstI digestion, whereas the 195-bp product generated from other CYP2C19 alleles remained uncut (see fig. 2).

PCR-RFLP mismatch test for CYP2C19*4. A 195-bp fragment of exon 1 was amplified by PCR, followed byPstI digestion and electrophoresis. The 195-bp PCR product is not digested in an individual (no. 22042) homozygous for the normal gene (CYP2C19*1). The restriction enzyme PstI completely digests the 195-bp PCR product in the CYP2C19*4allele to yield 167- and 28-bp restriction fragments as shown in the positive control (P). Four DNA samples were heterozygous for theCYP2C19*4 allele showing a digested 167-bp fragment: CE (CYP2C19*4/CYP2C19*1; EM), no. 32006 (CYP2C19*2/CYP2C19*4; PM), no. 32125 (CYP2C19*4/CYP2C19*1; EM) and CB (CYP2C19*2/CYP2C19*4; PM). The sizes of the molecular weight markers (M) are shown on the left.
As a positive control for CYP2C19*4, exon 1 of subject CE was amplified by PCR as described above. The products were cloned into the pCRII TA cloning vector (Invitrogen, San Diego, CA), transformed into E. coli DH5α (Life Technologies, Gaithersburg, MD), and a clone containing the CYP2C19*4 defect selected by sequencing. This clone was reamplified by PCR, and 0.5 ng of gel-purified DNA was utilized in subsequent PCR tests.
Expression of CYP2C19 in yeast, Western blot, and Northern blot analyses.
The cloning of wild-type CYP2C19*1A cDNA in pAAH5 has been described (Romkes et al., 1991). This cDNA was cloned in the HindIII site of pUC(pUC2C19). A modified CYP2C19 cDNA containing a GTG as the initiation codon was constructed by replacing the 5′ HindIII-SacI fragment of pUC2C19 with a PCR product generated by a mutant forward primer (5′-GCGAAGCTTAAAAAA GTG GATCCTT-3′) and the reverse primer (5′-GCAAGCTTGCCAGACCATCTGTGCTTCT-3′). The modified cDNA insert was retrieved with HindIII, subcloned into pAAH5 and verified by sequencing.
The protease-deficient Saccharomyces cerevisiae strain 334 was transformed with the wild-type and mutant 2C19 plasmids in pAAH5 and Leu+ transformants selected as described previously (Faletto et al., 1992). Ten colonies from each transformation were grown to mid-log phase, and Western blot analysis performed on cell lysates prepared from 3 × 107 cells with a primary polyclonal antibody to CYP2C9 which recognizes CYP2C9, CYP2C18 and CYP2C19 on Western blot analysis as described previously (Goldstein et al., 1994). The antibody was a generous gift from Dr. Jerome Lasker, Mount Sinai School of Medicine. Total RNA (25 μg) isolated from these cultures (Ausubelet al., 1990) was analyzed by Northern blot analysis with a32P-labeled CYP2C19 cDNA probe which consisted of the complete CYP2C19 cDNA excised from pAAH5 (Romkes et al.,1991).
Coupled in vitro transcription/translation assay.
CYP2C19*1A cDNA was amplified by PCR with the forward primer (5′-GCGGATATCGAATTCGGGCTTCAGTGGATCCTT-3′) and reverse primer (5′-GTAAGTCAGCTGCAGTGATTA-3′). The PCR product was digested withEcoRV and SacI and ligated into similarly digested CYP2C19 in Bluescript. The resultant plasmids containing the wild-type and the modified GTG codon were digested withHindIII and BglII, and the inserts ligated into the pSP64 poly(A) vector (Promega, Madison, WI) which was digested with HindIII and BamHI. Plasmids were propagated in Escherichia coli DH5α and sequences confirmed by DNA sequencing.
Plasmid templates (1 μg) were transcribed with SP6 RNA polymerase and translated with rabbit reticulocyte lysate in the presence of35S-methionine with the coupled in vitro system (Promega, Madison, WI). The reaction products were separated on 4 to 20% gradient polyacrylamide gels (Novex, San Diego, CA) under denaturing and reducing conditions. The gel was soaked in isopropanol/water/acetic acid (25:65:10) and then in Amplify scintillant (Amersham, UK), dried and exposed to X-ray film. Recombinant yeast microsomes containing CYP2C19 were electrophoresed on the same gel, and a portion of the gel was subjected to Western blot analysis to verify the size of the translated protein.
Data analysis.
HI values of 62 of the 234 individuals in the European population were excluded because of either intake of drugs known to show a metabolic interaction with mephenytoin (five subjects), missing HI values (21 subjects) or recovery of <10% of the administered dose of both dextrorphan and 4′-hydroxymephenytoin (36 subjects). HI values were thus considered reliable for 172 subjects. Blood samples were available for genotyping analysis of 173 of the 234 subjects. Both HI indices and genotype analysis were available for a total of 130 individuals and were analyzed by frequency distribution. Frequencies and confidence limits for the CYP2C19 alleles were calculated by methods described by Hahn and Meeker (1991) with all 173 individuals who were genotyped (346 alleles).
Results
An American subject CE was predicted to be an EM by a HI of 1.68 and S/R ratio of 0.28 which increased to 15 after acidification of the urine. This subject was thought to be homozygous for CYP2C19*1B, and the subject’s DNA was sequenced to verify sequence differences between two wild-type alleles, which we are terming CYP2C19*1A(Romkes et al., 1991) and CYP2C19*1B (Richardsonet al., 1995; S. M. F. deMorais, J. Blaisdell and J. A. Goldstein, unpublished data). Both alleles have been found to metabolize mephenytoin in cDNA expression systems (Goldsteinet al., 1994; Richardson et al., 1995).CYP2C19*1B differs from CYP2C19*1A by an A → G substitution at base pair 991 resulting in the amino acid change I330V (Romkes et al., 1991; Richardson et al., 1995) and a C → T noncoding change at base pair 99 (S. M. F. deMorais, J. Blaisdell and J. A. Goldstein, unpublished data). Although preliminary sequencing suggested that CE was homozygous forCYP2C19*1B, subsequent sequencing of all exons and exon-intron junctions revealed that CE was heterozygous for an A → G mutation at the first base of exon 1, changing the initiation codon from ATG → GTG (fig. 1). The sequence of the remainder of both alleles was identical with that ofCYP2C19*1B. The new allele containing an altered initiation codon was termed CYP2C19*4.

Sequence of the CYP2C19*4 defect. Antisense sequences of exon 1 of the normal (CYP2C19*1) and a new defective allele (CYP2C19*4) shown in the panels are from an automated sequencer (Applied Biosystems, Foster City, CA). The sense sequences are shown below the panels: (A) an EM of mephenytoin (CE) who is heterozygous for the CYP2C19*4 defect; (B) an EM who is homozygous for the normal ATG at this position. An arrow identifies the G → A mutation at the first base of the initiation codon (top) versus the normal ATG (bottom).
The CYP2C19*4 defect creates a Eco57I restriction endonuclease site in exon 1, and this feature was used in the development of a PCR-RFLP CYP2C19*4 genotyping test (fig.2). The 285-bp PCR product of exon 1 is undigested in individuals homozygous for the normal ATG (CYP2C19*1A or CYP2C19*1B). Eco57I completely digests the subcloned CYP2C19*4 product, producing 186- and 99-bp fragments. CE was heterozygous for theCYP2C19*4 allele showing all three bands.
The frequency of the three defective CYP2C19 alleles was determined in 173 European Caucasians. Phenotyping data (HI values) were available for 130 of these subjects. The frequency distribution of the HI values indicated that the antimode separating EMs from PMs was not unequivocal (fig. 3). However, comparison of HI values, genotypes, and S/R ratios before and after acidification indicated that HI values ≥40 have approximated the PM phenotype (table 1). Among eight putative PMs available for genotyping, six were explained by theCYP2C19*2 allele. One PM (no. 32006; HI = 47) was heterozygous for CYP2C19*2 and CYP2C19*4 (fig.2). One putative PM (no. 32080; HI = 43) remained unexplained as a possible outlier. One PM in table 1 was not available for phenotype and is not included in figure 3. Only one additional CYP2C19*4allele was found out of 173 individuals (an unphenotyped individual no. 32125) (fig. 2). The frequency of CYP2C19*4 was 2 of 346 alleles or ∼0.006 (0.002–0.021, 95% confidence interval) in this Caucasian population. The frequency of CYP2C19*2 was 0.13 (0.10–0.17, 95% confidence interval (46 of 346 alleles). Only oneCYP2C19*3 allele was detected in 346 alleles with a frequency of 0.003 (0.001–0.016, 95% confidence limits). The frequency of PMs was 9 of 172 phenotyped individuals (5.2% with 95% confidence limits of 2.8–9.7%).

Distribution of HIs determined in 130 genotyped Caucasian European subjects. Genotypes are indicated by shaded bars. An arrow indicates that individuals with a HI ≥ 40 were considered to be putative PMs of mephenytoin. m1, CYP2C19*2; m2,CYP2C19*3; m3, CYP2C19*4; wt, EM alleles (CYP2C19*1A and CYP2C19*1B).
Genotypic and phenotypic data on European subjects with HI > 201-a
Five of 29 additional PMs of mephenytoin previously genotyped by our laboratory (de Morais et al., 1994a, b; Sarich et al., 1997; Brøsen et al., 1995; Balian et al., 1995; Goldstein et al., 1997; J.A. Goldstein, J. Blaisdell, C. Beyeler and A.K. Daly, unpublished data) were outliers whose PM phenotype could not be completely explained by theCYP2C19*2 and CYP2C19*3 alleles. Reanalysis of all outliers for CYP2C19*4 in the present study revealed that one Canadian PM (Caucasian subject CB) was heterozygous forCYP2C19*2/CYP2C19*4 which was verified by sequencing (data not shown). This genotype is consistent with the PM phenotype (Sarichet al., 1997) based on the recovery of <1% of the dose of mephenytoin in the urine as 4′-hydroxymephenytoin and urinary S/R ratios of 1.04 before and 1.09 after acidification. Thus, theCYP2C19*4 allele accounted for two out of 74 alleles in 37 Caucasian PMs (∼3% of the PM alleles).
To verify that CYP2C19*4 represents a defective allele, the initiation codon of CYP2C19*1A (Romkes et al., 1991) was mutated to GTG, and both cDNAs were expressed in a yeast cDNA expression system. Ten recombinant yeast colonies from each transformation were analyzed by Western blot analysis (fig.4). Nine of 10 yeast colonies transformed with the wild-type 2C19 cDNA expressed recombinant CYP2C19 protein (fig. 4). None of the yeast colonies transformed with the mutant cDNA expressed the CYP2C19 protein. However, Northern blots showed comparable amounts mRNA in colonies transformed with the two cDNAs, which suggests that the differences in protein expression occurred at the level of translation (fig. 5).

Western blots of recombinant CYP2C19 in yeast transformed with CYP2C19 cDNA containing an ATG or GTG initiation codon. Each lane (1–10) contains cell lysate prepared from cultures grown from a single yeast colony transformed with either 2C19 cDNA containing an ATG or GTG as an initiation codon. Recombinant CYP2C9 (2 pmol) and CYP2C19 (3 pmol) expressed in yeast microsomes are shown for comparison. Immunodetection was achieved with a primary polyclonal antibody to CYP2C9. An arrow marks the position of CYP2C19.

Northern blots of recombinant CYP2C19 in yeast transformed with CYP2C19 cDNA containing an ATG or GTG initiation codon. Each lane (1–10) contains total RNA (25 μg) isolated from the transformed yeast colonies described in figure 4 or control yeast (334). Blots were subjected to Northern analysis and hybridized with a32P-labeled CYP2C19 cDNA probe.
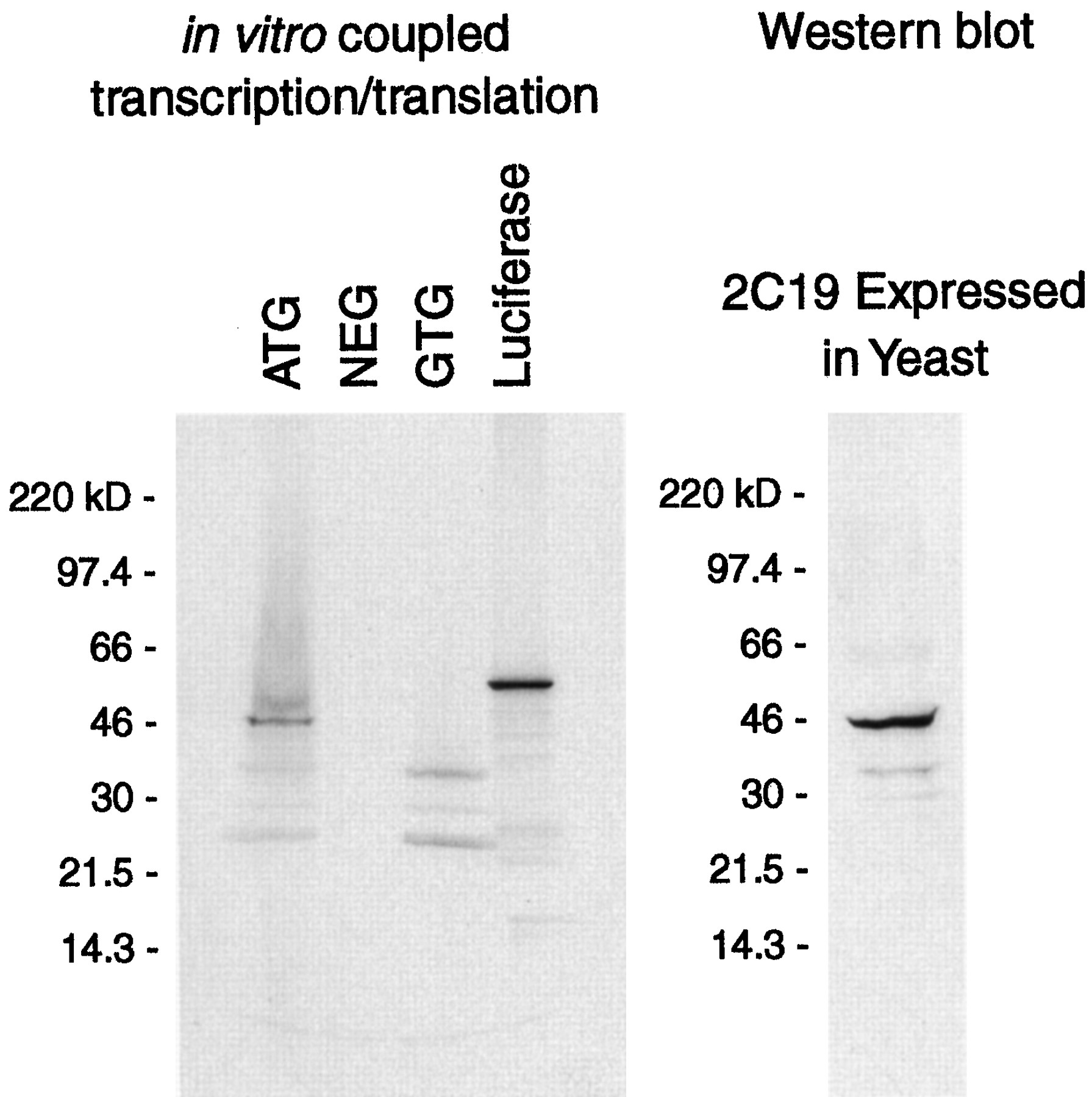
CYP2C19 cDNAs containing either an ATG or a GTG as the initiation codon were also used as templates in a coupled in vitrotranscription/translation assay (fig. 6). In three experiments, a protein with the approximate size of CYP2C19 (46 kd) was transcribed and translated from the ATG construct, whereas the protein was not translated from the GTG template.

Coupled in vitro transcription/translation assay using CYP2C19 cDNA templates containing either an ATG or GTG initiation codon. (left panel) Plasmid templates containing native 2C19 cDNA (ATG), mutant 2C19 cDNA (GTG), luciferase cDNA or vector without insert (NEG) were used as templates for the coupled in vitrotranscription/translation assay as described under “Materials and Methods.” (right panel) Recombinant CYP2C19 (2 pmol) expressed in yeast microsomes was electrophoresed on the same gel and subjected to Western blot analysis with a polyclonal antibody to CYP2C9 to verify the appropriate size of the correctly translated CYP2C19 protein.
Discussion
Mutations of CYP2C19 are responsible for a well-known polymorphism in drug metabolism in humans which affects the metabolism of the anticonvulsant agent mephenytoin as well as that of other drugs. Recently, two mutations, CYP2C19*2 and CYP2C19*3have been shown to account for >99% of Oriental but only ∼88% of 37 Caucasian PM alleles, which suggests that other defective alleles contribute to the PM phenotype in Caucasians (de Morais et al., 1994a, b; Sarich et al., 1997; Brøsen et al., 1995; Goldstein et al., 1997; J. A. Goldstein, unpublished data). The present study identifies a new allele, CYP2C19*4, which contributes to the PM phenotype in Caucasians. This new allele contains an ATG → GTG mutation in the initiation codon.
The defective nature of the CYP2C19*4 allele is shown by the fact that two Caucasian PMs were heterozygous forCYP2C19*2/CYP2C19*4. Although the frequency ofCYP2C19*4 is only 0.6% in Caucasians, this allele accounted for the phenotype of 2 of 37 Caucasian PMs. The genetic tests forCYP2C19*2, CYP2C19*3 and CYP2C19*4 now correctly identify ∼91% of the defective alleles in 37 Caucasian putative PMs studied in our laboratory (de Morais et al., 1994a, b;Sarich et al., 1997; Brøsen et al., 1995;Goldstein et al., 1997; and present studies) and predict the phenotype in 86% of these PMs. This may represent an underestimate of the efficiency of the tests because of the possibility of noncompliance in urinary collection and the fact that certain individuals are not available for re-phenotyping.
This the first example of the utilization of a GTG as the initiation codon in CYP proteins. However, several precedents exist for the initiation of translation from a GTG codon for certain other proteins in bacteria and Drosophila (Shaw and Fulco, 1992; Sugiharaet al., 1990), although this codon is often translated with reduced efficiency in these systems. In mammalian systems, a GTG mutation was reported in the initiation codon of the gene which codes for a growth factor protein in two Japanese individuals with Norrie disease (an X-linked recessive disease characterized by bilateral congenital blindness) (Isashiki et al., 1995), and a GTG mutation was also reported in the initiation codon of the gene encoding the G protein of adenylate cyclase in Albright’s hereditary osteodystrophy (Patten et al., 1990). However, the role of the GTG codon in affecting the efficiency of translation of these genes was not investigated.
Mutation of the initiation codon of CYP2C19 cDNA to a GTG in the present study abolished expression of recombinant CYP2C19 in a eukaryotic yeast cDNA expression system, confirming the defective nature of the CYP2C19*4 allele. Because CYP2C19 mRNA levels were similar in yeast transformed with the two cDNAs, these data suggest that the GTG codon produces a marked decrease in the translation of CYP2C19. Results of an in vitrotranscription/translation assay also suggest that the GTG codon interferes with translation.
CYP2C19*3 represents ∼25% of the PM alleles in Orientals, but it is rare in Caucasians (de Morais et al., 1994a, b;Brøsen et al., 1995; Goldstein et al., 1997). We previously found this allele in 1 of 11 Caucasian PMs from Denmark, but it was not present in 105 American-Caucasians or 97 Saudi Arabians. The present study verifies the presence of this allele in a European Caucasian population with a low frequency of 0.003 (95% confidence limits, 0.001–0.016).
In summary, the present study reports a new PM allele ofCYP2C19, in which the initiation codon is changed from an ATG to a GTG. This is the first example of a GTG codon as the initiation codon of a CYP protein. The defective nature of this allele was verified by its lack of expression in a yeast cDNA expression system, the absence of translation in an in vitro coupled transcription translation assay, and its presence in two previously unexplained PMs of mephenytoin. This mutation accounted for 3% of 37 Caucasian PM alleles, increasing the efficacy of the genetic tests in predicting the PM phenotype to ∼86% in Caucasians.
Acknowledgments
The authors thank Richard W. Morris, of Analytical Sciences, Inc., Research Triangle Park, NC for his expert statistical analyses.
Footnotes
-
Send reprint requests to: Dr. Joyce Goldstein, NIEHS, PO Box 12233, Research Triangle Park, NC 27709.
-
↵1 This work was supported in part (S.M., C.B., P.D.) by the Swiss Cancer League, Switzerland (FOR063); League against Cancer of Fribourg, Switzerland (FOR381.88); Cancer Research, Switzerland (AKT617); and Fund for Clinical Research against Cancer, Gustave-Roussy Institute, Villejuif, France (88D28), by USPHS grants GM31304 (G.R.W.); and by Program Project Grant 32165, National Institutes of Health (T.C.S., J.M.W.).
-
↵2 Current affiliation: University of Florida Shands Cancer Center, Health Science Center, PO Box 100286, Gainesville, FL 32610-0286.
-
↵3 Current affiliation: Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT.
-
↵4 The present paper uses a nomenclature for theCYP2C19 alleles based on the recommendations of Dalyet al. (1996). We denote the wild-type allele reported byRomkes et al. (1991) as CYP2C19*1A. A second wild-type allele (C99T, A991 G, Ile331 Val) (Richardson et al., 1997; S.M.F. de Morais, J. Blaisdell and J. A. Goldstein, unpublished data) is designated CYP2C19*1B. The mutant alleles previously reported by our laboratory (deMorais et al., 1994a, b) are designated as follows: CYP2C19m1 is designatedCYP2C19*2, CYP2C19m2 is designatedCYP2C19*3 and CYP2C19m3 is designatedCYP2C19*4.
- Abbreviations:
- PM
- poor metabolizer
- EM
- extensive metabolizer
- PCR
- polymerase chain reaction
- HI
- hydroxylation index
- CYP
- cytochrome P450
- RFLP
- restriction fragment length polymorphism
- Received March 25, 1997.
- Accepted August 21, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}